Objectifs pédagogiques
- Connaître les principaux arguments du diagnostic de sclérose en plaques.
- Connaître les principes de prise en charge.
Hiérarchisation des connaissances
I. Pour comprendre
- La SEP est une affection inflammatoire chronique du système nerveux central (SNC) surve- nant chez l'adulte jeune.
- Le diagnostic repose sur les notions de dissémination spatiale (au moins deux territoires neurologiques touchés) et de dissémination temporelle (au moins deux épisodes neuro- logiques séparés d'au moins 1 mois). Ces deux critères peuvent être apportés par des données cliniques et/ou IRM.
- La ponction lombaire (PL) contribue au diagnostic positif et au diagnostic différentiel. Elle permet d'affirmer le caractère inflammatoire de l'affection, localisé au SNC. Elle peut rem- placer le critère de dissémination dans le temps s'il est absent.
- Les traitements ont pour but 1) d'accélérer la récupération des symptômes des poussées; 2) de limiter la fréquence des poussées et la progression de la maladie; 3) d'améliorer les symptômes résiduels.
II. Épidémiologie et étiologie
 Adulte jeune (20–40 ans), prédominance féminine (3 femmes pour 1 homme).
Adulte jeune (20–40 ans), prédominance féminine (3 femmes pour 1 homme).- Prévalence : 100000 patients en France (1 personne/1000). Gradient nord-sud : la SEP est plus fréquente quand on s'éloigne de l'Équateur (deux fois plus de SEP dans les pays scandinaves que méditerranéens).
- La SEP est une maladie multifactorielle :
- facteurs génétiques : population caucasoïde, concordance 30 % chez les jumeaux monozygotes contre 2 à 3 % chez les hétérozygotes, gènes de susceptibilité (liés en particulier au groupe HLA);
- facteurs d'environnement : infections virales (EBV), hygiène (une moindre exposition à certaines infections dans l'enfance pourrait augmenter le risque), parasitoses (effet protecteur), vitamine D (une carence est associée à une augmentation du risque de survenue de la maladie et peut-être à la sévérité de l'évolution), tabac (augmente le risque et aggrave l'évolution), exposition au tabac...
III. Physiopathologie
- Maladie inflammatoire chronique du système nerveux central (SNC). La gaine de myéline est la cible du système immunitaire via un clone lymphocytaire auto-réactif (c'est une maladie auto-immune à médiation cellulaire), mais il existe également une atteinte de l'axone (primitive et secondaire).
- Plaques de démyélinisation focales disséminées dans la substance blanche (principale zone myélinisée) du SNC (nerf optique, zones périventriculaires, corps calleux, cervelet, moelle épinière, etc.). Les symptômes cliniques des poussées sont liés à la localisation de la plaque, expliquant leur diversité. Remyélinisation possible par les oligodendrocytes, expliquant la récupération des poussées. Les mécanismes de la phase progressive associent une inflammation plus diffuse dans le SNC et une neuro-dégénérescence.
IV. Clinique
A. Présentation clinique
Elle est très variable, fonction de la localisation de la plaque. Elle peut être mono ou pluri-symptomatique, la multiplicité des symptômes ne signifiant pas multiplicité des lésions.
1. Les deux événements de base
Les symptômes neurologiques s'installent le plus souvent (85 à 90 % des cas) de manière subaiguë, en quelques heures ou quelques jours, puis régressent, définissant la poussée ; plus rarement, les symptômes sont insidieux d'emblée, s'aggravant sur plusieurs mois ou années (10 à 15 %).
a. La poussée
 Une poussée se définit comme l'apparition de nouveaux symptômes, la réapparition d'anciens symptômes ou l'aggravation de symptômes préexistants, s'installant de manière subaiguë en quelques heures à quelques jours, et récupérant de manière plus ou moins complète. Sa durée est au minimum de 24 heures. Une fatigue seule, ou des symptômes survenant dans un contexte de fièvre, ne sont pas considérés comme une poussée. Par définition, deux poussées doivent être séparées d'au moins 1 mois.
Une poussée se définit comme l'apparition de nouveaux symptômes, la réapparition d'anciens symptômes ou l'aggravation de symptômes préexistants, s'installant de manière subaiguë en quelques heures à quelques jours, et récupérant de manière plus ou moins complète. Sa durée est au minimum de 24 heures. Une fatigue seule, ou des symptômes survenant dans un contexte de fièvre, ne sont pas considérés comme une poussée. Par définition, deux poussées doivent être séparées d'au moins 1 mois.
b. La progression
La progression est définie comme l'aggravation continue, sur une période d'au moins 6 mois, de symptômes neurologiques. Une fois commencée, elle ne s'interrompt plus et est donc une cause majeure de handicap chez les patients atteints de SEP.
2. Formes cliniques
L'évolution globale de la SEP est polymorphe, reflet de l'interaction entre les poussées et la progression continue du handicap. Trois formes cliniques principales de SEP peuvent être défi- nies en fonction de la combinaison de ces deux événements de base.
a. Forme rémittente-récurrente
La forme rémittente-récurrente est composée exclusivement de poussées qui peuvent laisser des séquelles, ces séquelles restant stables entre deux épisodes. Elle débute vers 30 ans en moyenne et représente 85 % des formes de début.
b. Forme secondairement progressive
La forme secondairement progressive est l'évolution naturelle tardive de la forme précédente, une phase de progression succédant à la phase rémittente. Elle peut toucher tous les patients initialement rémittents, après une période plus ou moins longue, de 15 à 20 ans en moyenne.
c. Forme primaire progressive
La forme primaire progressive ou progressive d'emblée, où la progression est présente dès le début, sans poussée, affecte 15 % des patients et débute en moyenne un peu plus tardivement, vers 40 ans. Elle se caractérise habituellement par une atteinte médullaire (limitation progressive du périmètre de marche par paraparésie spastique).
3. Les symptômes
Les symptômes dépendent de la phase clinique.
a. Les poussées inaugurales
La première poussée de la maladie dans les formes rémittente-récurrente correspond le plus souvent à un des tableaux suivants :
- myélite partielle, caractérisée par des troubles sensitifs des membres, de topographie médullaire, parfois associés à des troubles moteurs et/ou sphinctériens. Les troubles sensi- tifs correspondent à des picotements, des fourmillements, des sensations d'hypoesthésie ou même d'anesthésie, des douleurs, des décharges, des sensations de striction ou d'étau, de ruissellement, de chaud, de froid. Le signe de Lhermitte est très évocateur : il s'agit d'une impression de décharge électrique très brève le long de la colonne vertébrale, parfois des membres, se déclenchant électivement à la flexion de la tête vers l'avant. Il reflète une démyélinisation des cordons postérieurs de la moelle cervicale ;
- la lésion médullaire, ovalaire, mesurant moins de trois vertèbres de hauteur et occupant moins de la moitié de la largeur de la moelle en axial, est visualisable sur l'IRM médullaire ;
- la névrite optique rétrobulbaire révèle la maladie dans un quart des cas. Baisse de l'acuité visuelle s'installant sur quelques heures à jours, associée à une douleur périorbitaire dans 80 % des cas, favorisée par la mobilisation du globe oculaire. Un scotome et une dyschro- matopsie rouge-vert sont souvent retrouvés. Le fond d'œil est normal au début, mais dans 10 % des cas, il est le siège d'un œdème papillaire. Une décoloration de la papille peut être observée dans les semaines qui suivent l'épisode aigu. La récupération de la fonction visuelle est complète dans 80 % des cas en 6 mois. Le phénomène d'Uhthoff se manifeste, après récupération de la névrite optique, par une baisse transitoire de quelques minutes de l'acuité visuelle, survenant à l'effort ou lors de l'augmentation de la température corporelle. La névrite optique, notamment lors d'un premier épisode, est une urgence dia- gnostique. En effet, si dans la SEP le pronostic est en général bon, il ne faut pas méconnaître certains diagnostics différentiels dont le pronostic est beaucoup plus péjoratif (névrite optique de la neuromyélite optique par exemple, ou baisse d'acuité visuelle dans le cadre d'une hypertension intracrânienne). Le bilan à réaliser en urgence comprend un examen ophtalmologique avec mesure de l'acuité visuelle (on évaluera la sévérité de l'atteinte visuelle), du champ visuel (on recherchera un scotome et son étendue), fond d'œil (on recherchera un œdème papillaire). Les potentiels évoqués visuels confirmeront dans un second temps la démyélinisation du nerf optique (allongement de la latence du P100) (figure 14.1). L'IRM des nerfs optiques peut également permettre de visualiser la lésion inflammatoire responsable de la baisse d'acuité visuelle (figure 14.2).
- Moins souvent le tableau initial correspond à un des tableaux suivants :
- atteinte du tronc cérébral (vertige par syndrome vestibulaire central ; diplopie par paralysie oculomotrice) ;
- atteinte du cervelet (ataxie);
- atteinte hémisphérique (atteinte sensitive et/motrice unilatérale);
- syndrome multifocal.
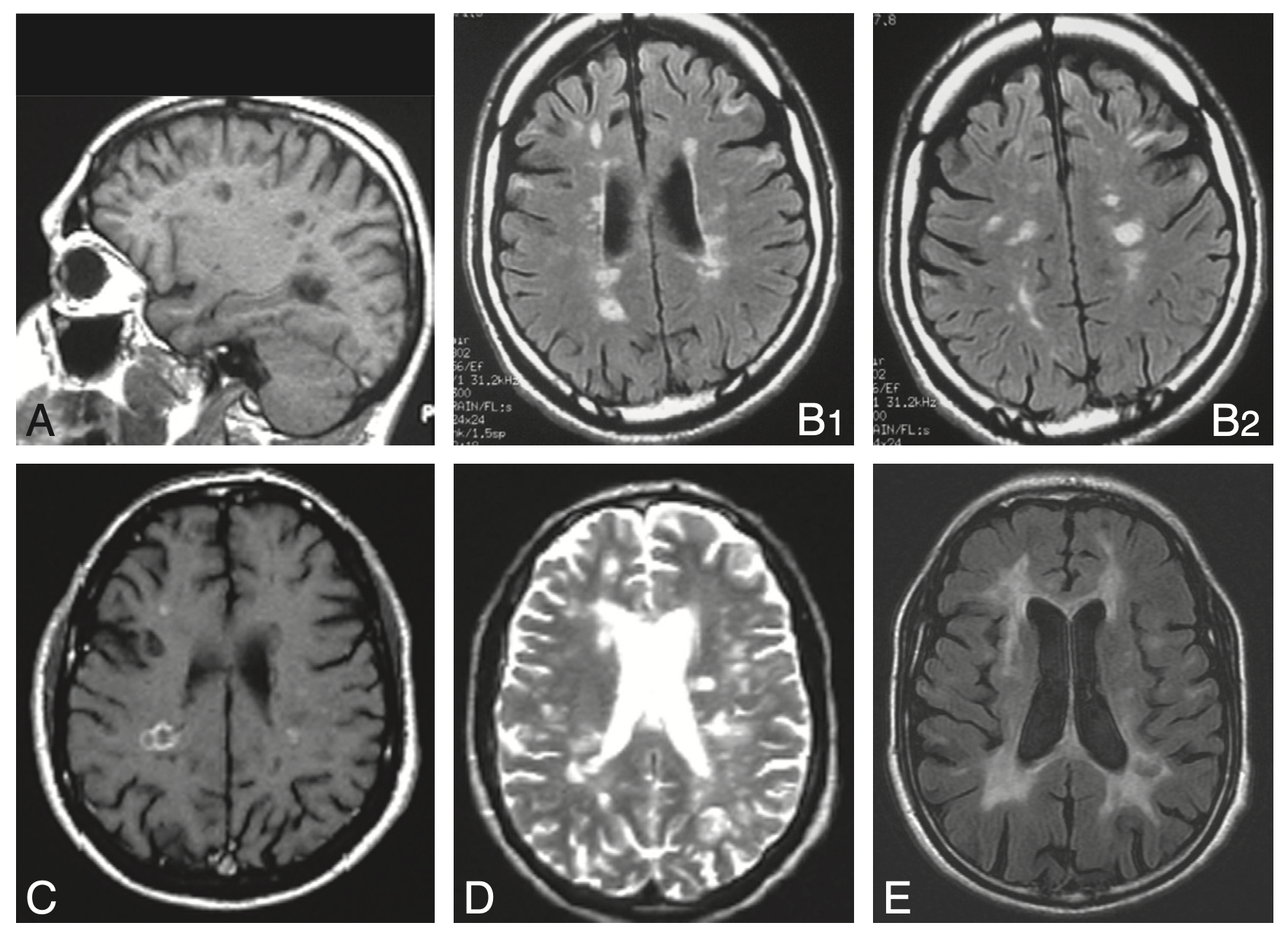
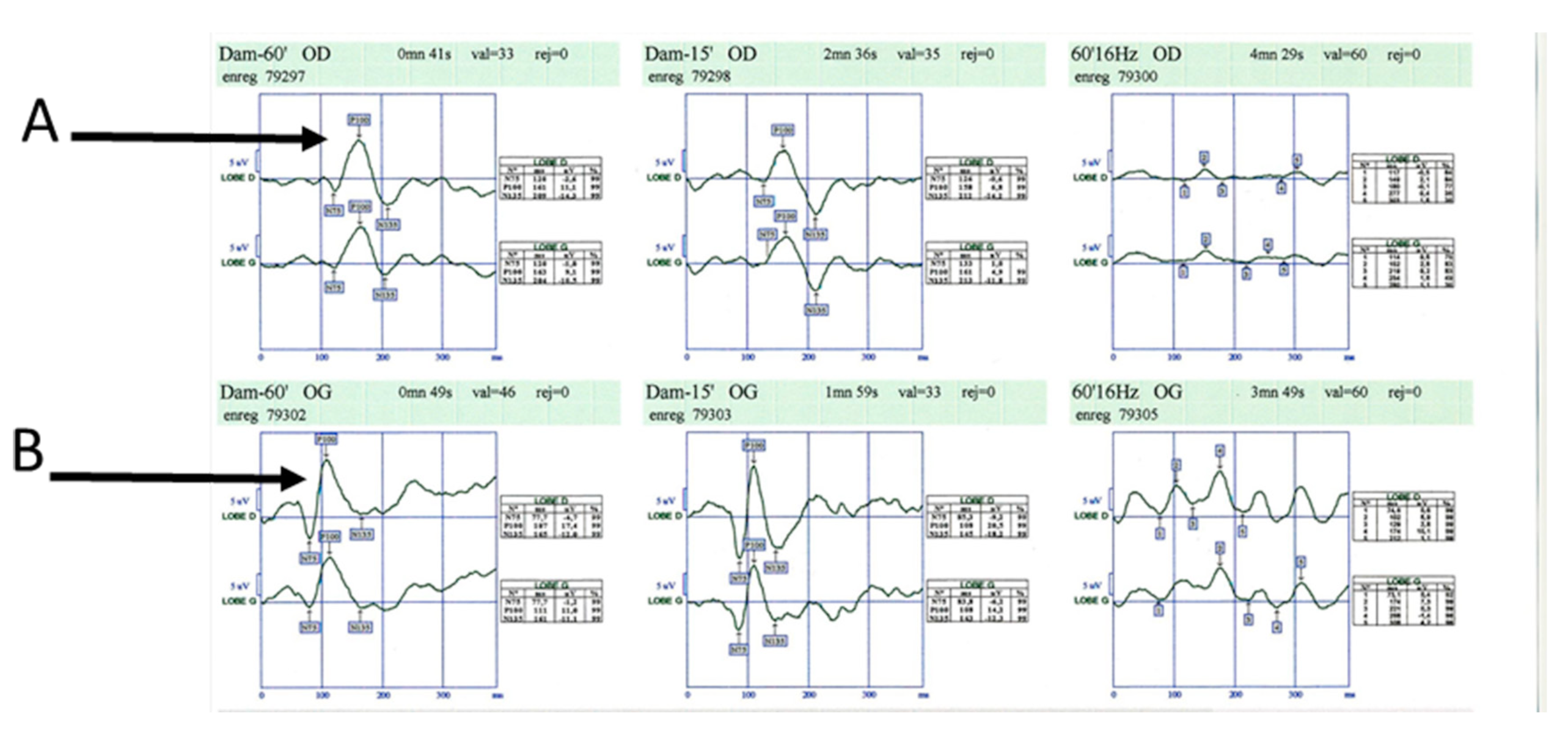
Fig. 14.1.  Potentiels évoqués visuels. Latence de l'onde P100 augmentée à droite (A) par rapport à la gauche (B).
Potentiels évoqués visuels. Latence de l'onde P100 augmentée à droite (A) par rapport à la gauche (B).
(Source : auteur.)
b. Les poussées ultérieures
Elles peuvent correspondre à n'importe lequel des tableaux décrits ci-dessus.
c. Les symptômes de la phase progressive
Ils sont dominés par les tableaux de paraparésies spastiques qui représentent le mode de début habituel des formes progressives primaires (troubles de la marche d'apparition insidieuse) mais des tableaux d'ataxies progressives sont possibles.
d. Les autres symptômes
Ils sont plus rares au tout début de la maladie, mais sont fréquents à la phase d'état : atteintes du tronc cérébral (ophtalmoplégie internucléaire, paralysie faciale centrale ou périphérique, névralgie faciale, dysarthrie, troubles de déglutition), troubles sphinctériens (hyperactivité vésicale responsable d'impériosités mictionnelles, pollakiurie; hypertonie sphinctérienne responsable d'une dysurie), fatigue, troubles cognitifs, douleurs neuropathiques...
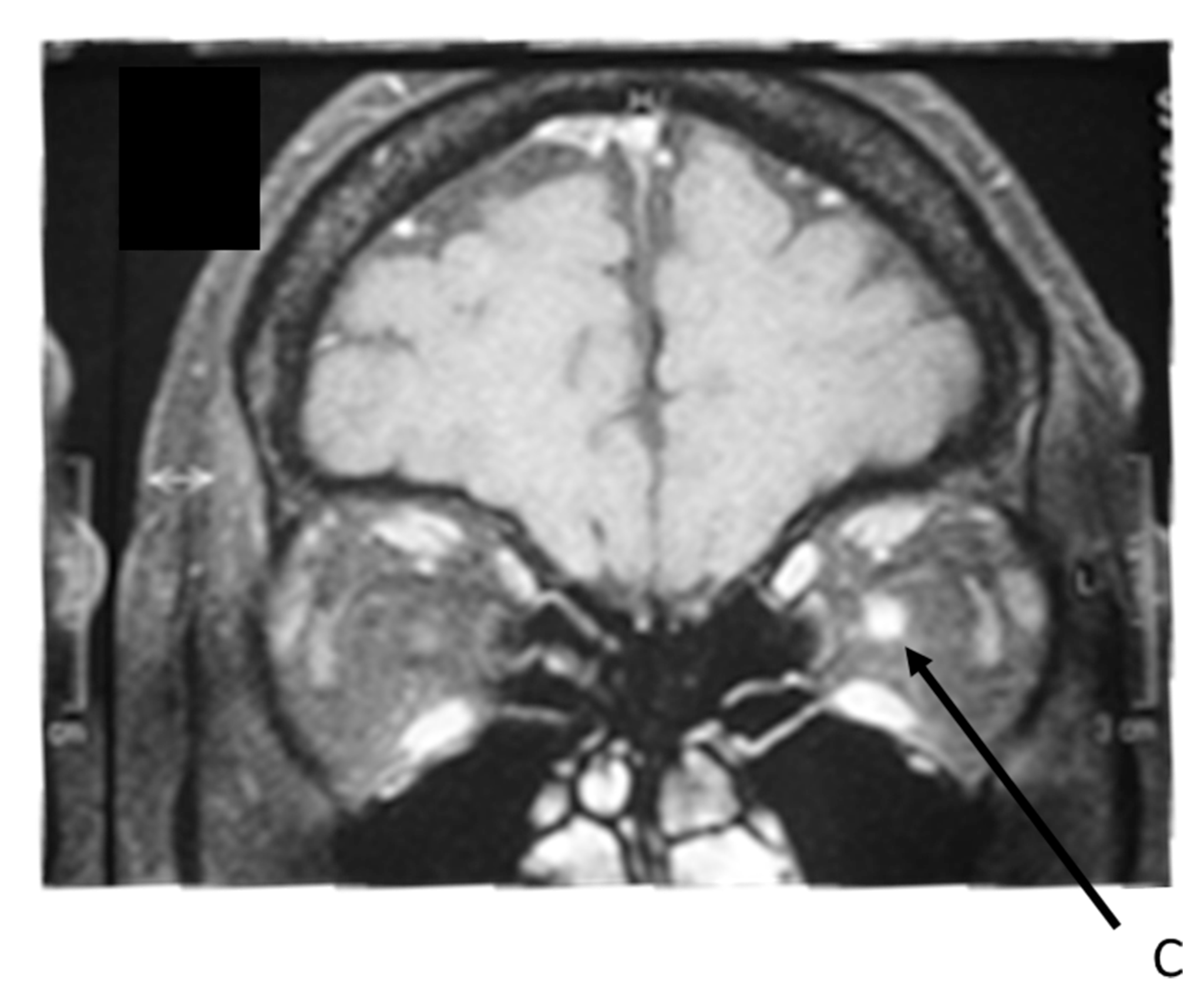
Fig. 14.2. IRM cérébrale et orbitaire en coupe coronale T1 avec injection de gadolinium. Hypersignal du nerf optique gauche.
(Source : auteur.)
B. Évolution
1. Le pronostic général
 On définit également aujourd'hui une maladie comme active s'il y a eu, au cours de l'année précédente, une poussée (activité clinique) et/ou de nouvelles lésions en séquences pondérées en T2 ou des lésions réhaussées par le gadolinium en séquences pondérées en T1 (activité radiologique).
On définit également aujourd'hui une maladie comme active s'il y a eu, au cours de l'année précédente, une poussée (activité clinique) et/ou de nouvelles lésions en séquences pondérées en T2 ou des lésions réhaussées par le gadolinium en séquences pondérées en T1 (activité radiologique).
Le pronostic global de la SEP est très variable, allant de formes bénignes ou pauci-symptomatiques à des formes graves, entraînant rapidement un état grabataire et une dépendance complète. L'espérance de vie moyenne n'est en revanche que peu réduite. Avant l'ère des traitements, on considérait qu'un tiers des malades devrait un jour utiliser un fauteuil roulant, alors qu'un quart avait une évolution bénigne, compatible avec une vie personnelle et professionnelle quasi normale. Les autres patients, tout en restant autonomes, gardaient peu à peu des séquelles permanentes limitant leurs activités. L'évolution est moins souvent sévère depuis l'utilisation des thérapeutiques immunologiques.
Le pronostic est imprévisible pour un individu donné.
Quelques facteurs cliniques prédictifs d'évolution ont pu être mis en évidence. L'âge de début jeune, le mode rémittent, un long délai entre les deux premières poussées semblent plutôt de meilleur pronostic. À l'opposé, les SEP débutant après 40 ans, plutôt de type primitivement progressif avec une atteinte motrice initiale, sont de mauvais pronostic. L'importance de la charge lésionnelle en IRM au début de la maladie est un facteur de mauvais pronostic.
C. Diagnostic
Il n'existe pas de test permettant de faire le diagnostic de SEP. Celui-ci repose donc sur des critères, évoluant avec le temps et la meilleure connaissance de la maladie. Il reste cependant basé sur un faisceau d'arguments : dissémination des symptômes et/ou des lésions dans le temps et dans l'espace, inflammation du LCS, absence d'atteinte générale et absence de meilleure explication. La démarche diagnostique implique donc une phase d'élimination de diagnostics différentiels.
1. Diagnostic positif
- La dissémination temporelle des lésions se définit comme la succession d'épisodes neuro- logiques dans le temps (exemple : une névrite optique à un temps T puis un syndrome cérébelleux l'année suivante). Elle doit être recherchée systématiquement à l'interrogatoire. Elle peut être mise en évidence cliniquement (un intervalle minimal de 1 mois est requis de principe entre deux poussées) mais aussi à l'IRM, soit par l'apparition de nouvelles lésions sur des IRM successives, soit sur l'association de lésions prenant et ne prenant pas le contraste. Dans les formes progressives d'emblée, on considère par définition que le critère de dissémination dans le temps est rempli quand la maladie évolue et s'aggrave depuis plus d'un an.
- La dissémination spatiale des lésions correspond à l'atteinte de plusieurs zones du SNC. Elle peut être mise en évidence par les données cliniques (une névrite optique et un syndrome pyramidal ne peuvent pas être expliqués par une seule lésion) ou par les examens paracliniques, en particulier l'IRM.
 Devant un tableau clinique évocateur, l'IRM est l'examen de prédilection pour confirmer la suspicion d'atteinte inflammatoire du système nerveux central et éliminer les diagnostics différentiels. L'IRM médullaire sera ainsi le premier examen proposé devant une myélite, l'IRM des nerfs optiques devant une suspicion de neuropathie optique.
Devant un tableau clinique évocateur, l'IRM est l'examen de prédilection pour confirmer la suspicion d'atteinte inflammatoire du système nerveux central et éliminer les diagnostics différentiels. L'IRM médullaire sera ainsi le premier examen proposé devant une myélite, l'IRM des nerfs optiques devant une suspicion de neuropathie optique.
La démarche diagnostique face à un malade suspect de SEP comporte donc cette quête de la dissémination spatiale et temporelle, cliniquement d'abord, mais aussi par l'intermédiaire des examens paracliniques qui accélèrent la procédure. - L'IRM, encéphalique éventuellement complétée par une IRM médullaire, est l'examen de choix pour le diagnostic de SEP (figures 14.3. et 14.4). Les lésions apparaissent sous la forme d'hypersignaux de la substance blanche sur les séquences pondérées en T2 (le liquide cérébrospinal – LCS – apparaît blanc lors de ces séquences) ou, de préférence, en T2/FLAIR (fluid attenuation inversion recovery) qui est une séquence T2 avec inversion du signal du LCS, qui apparaît en hyposignal, permettant une meilleure différenciation avec les lésions. Elles peuvent aussi apparaître, inconstamment, en hyposignal («trous noirs») sur les séquences en T1. Les lésions sont ovoïdes, de plus de 3 mm habituellement, localisées majoritairement dans la substance blanche périventriculaire, avec un grand axe perpendiculaire à l'axe des ventricules. Elles peuvent également être juxta-corticales (au contact du cortex, ne respectant pas les fibres en U), sous-tentorielles (dans le cervelet ou le tronc cérébral) ou médullaires. Les lésions récentes (en pratique moins de 1 mois) apparaissent en hypersignal T1 après injection de produit de contraste (gadolinium). Une IRM médullaire peut être réalisée si la symptomatologie initiale évoque cette localisation ou si le nombre et la localisation des hypersignaux cérébraux ne sont pas suffisants pour poser le diagnostic.
- Les critères les plus récents, ceux de McDonald (2017), autorisent le diagnostic de SEP devant un tableau clinique typique, dès la première poussée, à condition qu'il y ait une dissémination spatiale (deux localisations différentes, soit cliniquement, soit dans deux des quatre zones stratégiques à l'IRM – périventriculaire, sous-tentorielle, juxta-corticale/corticale ou médullaire) et une dissémination temporelle (lésions d'âge différent objectivées par une prise de contraste pour certaines et pas pour d'autres). Dans le cas contraire, il faudra attendre une nouvelle poussée clinique ou de nouvelles lésions à l'IRM pour définir cette dissémination temporelle et/ou spatiale. En pratique, pour mettre cette nouvelle poussée en évidence précocement, on réalise souvent une nouvelle IRM environ 3 mois après l'IRM initiale. Ces critères de McDonald (2017) permettent aussi, pour affirmer le diagnostic plus précocement, de remplacer le critère de dissémination temporelle par la démonstration d'une inflammation du liquide cérébrospinal (présence de bandes oligoclonales seulement).
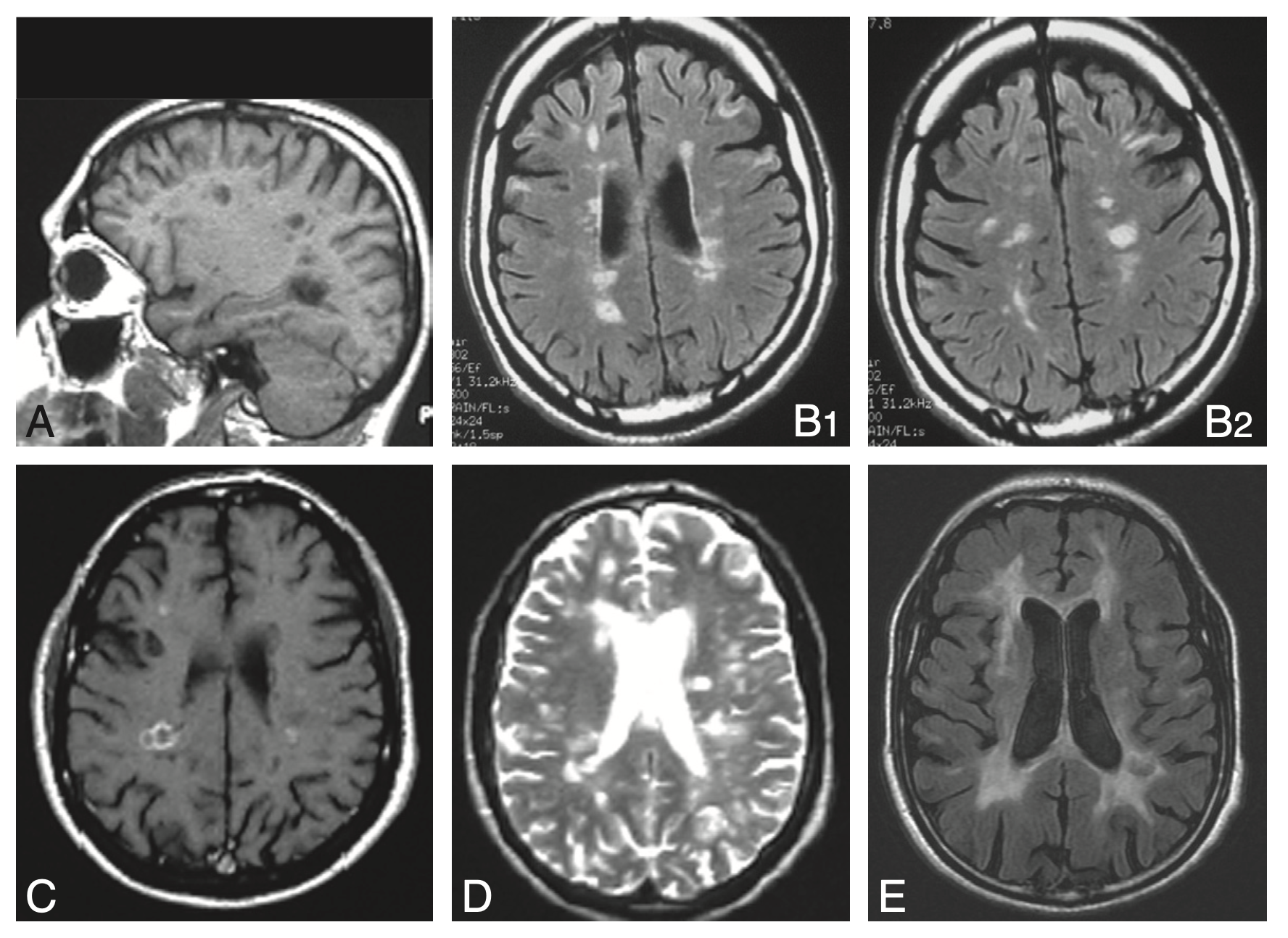
Fig. 14.3. Imagerie cérébrale dans la sclérose en plaques.
A. IRM en séquence T1 : « trous noirs » dans la substance blanche.
B. IRM en séquence FLAIR : hypersignaux à prédominance périventriculaire.
C. IRM en séquence T1 avec injection de gadolinium : lésions périventriculaires.
D. IRM en séquence T2 : hypersignaux à prédominance périventriculaire.
E. IRM en séquence FLAIR chez un patient ayant une maladie évoluée : hypersignaux confluents de la substance blanche associés à une atrophie corticale.
(Source : CEN, 2019.)
- L'analyse LCS permet de mettre en évidence l'inflammation du SNC. Le LCS est considéré comme inflammatoire s'il existe des bandes oligoclonales en iso-électrofocalisation (plus de 90 % des SEP) et/ou un index IgG augmenté (> 0,7) correspondant à une sécrétion intrathécale d'IgG. C'est la mise en évidence de cette synthèse intrathécale qui contribue aux critères du diagnostic. Les autres éléments ont moins de valeur diagnostique mais ils doivent alerter sur des diagnostics différentiels quand leur valeur n'est pas dans les normes attendues : la protéinorachie n'est augmentée que dans 25 % des cas en restant inférieure à 1 g/L. La cytorachie, composée d'éléments mononucléés (surtout des lymphocytes et plasmocytes), est supérieure à 4 éléments/mm3 dans un tiers des cas, mais le plus souvent inférieure à 50 éléments/mm3. Il existe une élévation des gammaglobulines dans le LCS dans 70 % des cas, alors qu'elles sont normales dans le sang. Le LCS peut aussi être normal. L'examen du LCS n'est pas systématique si les critères diagnostiques sont déjà remplis, mais il aide aussi à éliminer des diagnostics différentiels.
- Les potentiels évoqués (PE) sont de moins en moins pratiqués. Ils ne sont réalisés que dans les cas cliniquement compatibles avec le diagnostic de SEP, mais à IRM ou LCS non concluants ou bien lors d'un doute sur l'organicité des troubles. Ils permettent d'apporter un argument de dissémination dans l'espace quand il n'est pas présent mais ne rentrent pas en compte pour le diagnostic. Les PEV restent les plus utilisés compte tenu de la fréquence de la névrite optique.
Cependant aucun de ces examens ne constitue un marqueur spécifique de la maladie et leurs perturbations peuvent être retrouvées dans des maladies pouvant être confondues avec la SEP.
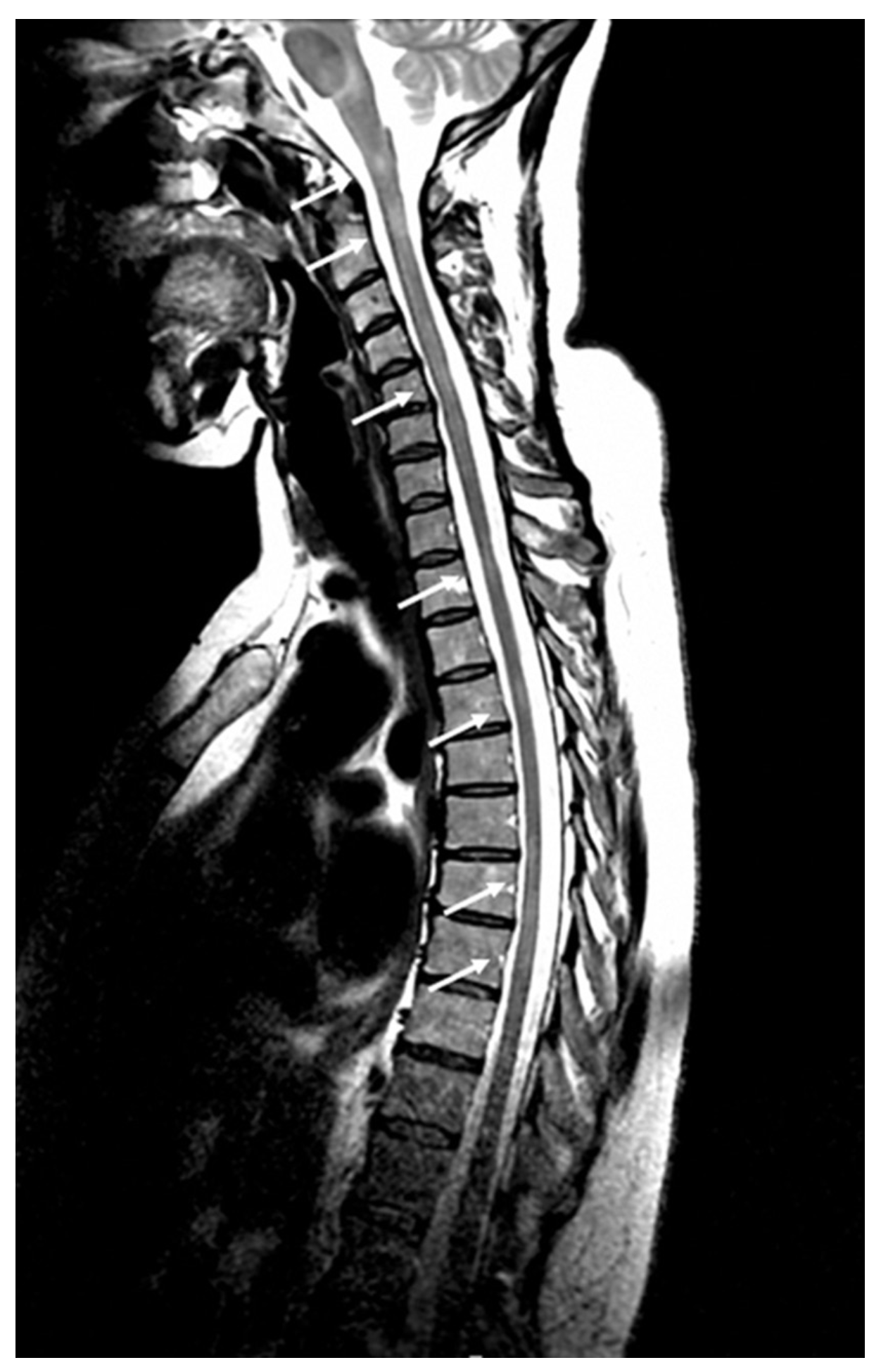
Fig. 14.4. Imagerie médullaire dans la sclérose en plaques.
IRM en séquence T2 : multiples hypersignaux le long de la moelle cervico-dorsale (flèches).
(Source : CEN, 2019.)
2. Diagnostic différentiel
Le principal diagnostic différentiel est représenté par les épisodes inflammatoires du SNC isolés sans dissémination spatiale et temporelle comme la névrite optique auto-immune idiopathique.
On a également mis en évidence d'autres affections inflammatoires limitées au SNC, comme la neuromyélite optique ou les maladies associées à des anticorps anti-MOG (Myelin Oligoden- drocyte Glycoprotein), où l'on retrouve des autoanticorps anti-système nerveux spécifiques. Chez l'enfant, l'encéphalite aiguë disséminée, maladie inflammatoire multifocale mais n'évoluant pas vers la chronicité, est un diagnostic différentiel.
Les maladies inflammatoires systémiques comme la sarcoïdose, la maladie de Behçet, le lupus érythémateux disséminé, la maladie de Gougerot-Sjögren, les artérites cérébrales, les infections à tropisme neurologique, les maladies cérébrovasculaires à attaques successives peuvent simuler une SEP rémittente. La recherche d'une altération de l'état général, d'une atteinte d'un autre organe que le SNC à l'interrogatoire ou à l'examen clinique général, la présence d'un syndrome inflammatoire dans le sang, d'une méningite et/ou l'absence de bandes oligoclonales dans le LCS sont des arguments pour évoquer ces maladies.
Les atteintes neurologiques localisées, même si elles évoluent par poussées, doivent faire rechercher une tumeur, une malformation vasculaire ou une compression médullaire. La neuro-imagerie éliminera ces diagnostics.
V. Traitements
A. Traitement de la poussée
Les corticoïdes à fortes doses permettent d'accélérer la récupération de la poussée. Ils sont prescrits en perfusion intraveineuse ou par voie orale à la dose de 1 g par jour pendant 3 jours (méthylprednisolone). Ils n'ont pas d'effet sur la prévention de nouvelles poussées. Leur utili- sation n'est pas systématique si les symptômes ne sont pas gênants.
B. de fond
 Ils ont pour but de réduire la fréquence des poussées et de ralentir la progression du handicap.
Ils ont pour but de réduire la fréquence des poussées et de ralentir la progression du handicap.
Les traitements de fond agissent tous sur la réponse immune soit de façon immunomodulatrice (en modifiant l'équilibre de certains systèmes immunologiques comme le réseau des cytokines), soit de façon immunosuppressive (en interférant avec le cycle cellulaire des cellules immunocompétentes ou en modifiant la distribution de ces cellules dans l'organisme).
Ils sont classés en traitements de première ligne (chez les patients naïfs de tout traitement ou sans critères de sévérité particuliers) et indiqués dans les formes actives de la maladie (caractérisées par la survenue d'une poussée et/ou de nouvelles lésions à l'IRM) ou de deuxième ligne, indiqués soit en cas de maladie très active (au moins deux poussées sévères au cours de la dernière année chez un patient sans traitement de fond), soit en cas d'échec (persistance d'une activité clinique et/ou radiologique) d'un autre traitement bien conduit.
Il est recommandé de traiter précocement, dès le diagnostic, pour ne pas laisser les lésions inflammatoires et le handicap résiduel s'accumuler, et de surveiller l'efficacité du traitement (absence de poussée, d'accumulation du handicap, absence de nouvelles lésions sur une IRM annuelle) pour proposer, si besoin, un changement de traitement.
C. Symptomatiques
Ils ont pour but de traiter les complications de la maladie, ce qui améliore la qualité de vie des malades. Ils représentent donc un complément essentiel des traitements précédents.
- La spasticité : peut être combattue par des antispastiques (baclofène ou dantrolène), à prescrire d'abord à faible dose pour éviter d'aggraver l'état moteur du malade par une hypotonie. Dans les spasticités sévères, les injections locales de toxine botulinique ou l'implantation d'une pompe intrarachidienne de baclofène peuvent être indiquées. La kinésithérapie permet de lutter contre l'hypertonie et les déformations.
- Les troubles urinaires doivent être dépistés et traités pour éviter une atteinte du haut appareil urinaire. En plus de l'interrogatoire, un bilan urodynamique et radiologique est souvent nécessaire. S'il existe une hyperactivité vésicale se traduisant par des urgences mictionnelles, les anticholinergiques sont utilisés. En cas de dysurie, les alphabloquants peuvent être prescrits. En cas de résidu post-mictionnel, la pratique d'autosondages intermittents pluri-quotidiens doit être proposée aux malades, éventuellement associée dans un second temps à des injections intravésicales de toxine botulinique. Les infections urinaires symptomatiques seront traitées pour éviter les pyélonéphrites. Elles constituent de plus des épines irritatives pouvant aggraver la spasticité.
- Les troubles sexuels, en particulier de l'érection, peuvent être améliorés par une prise en charge médicamenteuse et par un suivi sexologique ou psychothérapique.
- Les douleurs peuvent être soulagées par des antalgiques classiques, des tricycliques ou certains antiépileptiques.
- La fatigue est difficile à combattre. Les médicaments antiasthéniques sont peu efficaces (amantadine, modafinil). L'activité physique adaptée est recommandée, notamment le reconditionnement physique à l'effort.
- Une prise en charge psychothérapique peut être nécessaire, car il existe souvent un syndrome dépressif associé. Des antidépresseurs peuvent être proposés en cas de besoin.



