Objectifs pédagogiques
- Diagnostiquer les principales formes d'épilepsie de l'enfant et de l'adulte.
- Identifier les situations d'urgence.
- Connaître les principes de la prise en charge.
Hiérarchisation des connaissances
I. Définitions
 Distinguer les notions de crise d'épilepsie et de maladie épileptique.
Distinguer les notions de crise d'épilepsie et de maladie épileptique.
A. Les épilepsies
Survenue transitoire de signes et/ou de symptômes cliniques due à une activité neuronale cérébrale excessive et anormalement synchrone.
En pratique, ces signes ou symptômes cliniques peuvent comporter une altération de la conscience et/ou des signes moteurs et/ou des signes sensoriels et/ou des signes psychiques ou cognitifs, et/ou des signes neuro-végétatifs,
On distingue les crises d'origine généralisée des crises d'origine focale. Attention, les crises d'origine focale peuvent se propager et évoluer vers une crise secondairement généralisée. On parle de crise focale secondairement généralisée (figure 15.1).

B. épileptiques
Maladies cérébrales chroniques caractérisée par :
1/ la survenue d'au moins une crise épileptique ;
2/ une prédisposition durable à générer des crises ;
3/ ainsi que par leurs conséquences neurobiologiques, neuropsychologiques, sociales et psychiatriques.
En pratique, cette prédisposition cérébrale durable à générer des crises est établie par :
- la survenue d'au moins deux crises épileptiques cliniquement avérées non provoquées espacées d'au moins 24 heures;
- l'identification d'un syndrome épileptique;
- la survenue d'une crise épileptique cliniquement avérée non provoquée et un risque estimé de récidive supérieur à 60 % dans les 10 ans, soit au moins un élément pathologique retrouvé :
- présence d'anomalies paroxystiques à l'EEG,
- lésion cérébrale préexistante épileptogène à l'imagerie,
- trouble neurodévelopemental préexistant.

Fig. 15.1. Définition physiopathologique des crises d'origine focale et des crises d'origine généralisée.
(Source : CEN, 2019, illustration de Carole Fumat.)

II. Diagnostic positif des crises épileptiques
A. Crises généralisées
1. Deux grandes catégories de signes cliniques sont habituelles dans les crises généralisées
- Les signes moteurs : d'emblée bilatéraux et symétriques, ils peuvent être :
- toniques : contractions musculaires segmentaires des agonistes et antagonistes, soutenues ;
- cloniques : mouvements rythmiques soutenus résultant de secousses musculaires segmentaires répétitives et rythmiques;
- tonicocloniques : succession dans le temps d'une phase tonique et d'une phase clonique ;
- atoniques : interruption brève et soudaine du tonus de tout ou partie du corps ;
- myocloniques : mouvement isolé ou en courte salve résultant d'une contraction musculaire isolée ou en courte salve.
- Les troubles de conscience : de durée variable, quelques secondes au cours d'une absence à quelques minutes en cas de crise tonicoclonique.
2. Classification des crises généralisées
L'identification des signes cliniques principaux permet de classer les crises généralisées :
- avec signes moteurs au premier plan : comme par exemple les crises tonicocloniques, les crises myocloniques;
- avec altération de la conscience au premier plan : comme par exemple les absences typiques
3. Présentation clinique de quelques crises généralisées
a. Crise généralisée tonicoclonique
Elle se déroule en trois phases successives.
- La phase tonique (20 à 30 secondes) comporte : une vocalisation, une abolition de la conscience, une contraction tonique soutenue axiale et des membres (d'abord en flexion puis en extension), une apnée avec cyanose, des troubles végétatifs importants (tachycardie, augmentation de la tension artérielle, mydriase, rougeur du visage, hypersécrétion bronchique et salivaire), morsure latérale de langue possible.
- La phase clonique (20 à 30 secondes) comporte : des secousses bilatérales, synchrones, intenses, s'espaçant progressivement.
- La phase résolutive (ou post-critique) de quelques minutes qui comporte : une altération profonde de la conscience, une hypotonie généralisée avec possibilité d'une énurésie. La respiration reprend, ample, bruyante (stertor), gênée par l'hypersécrétion bronchique et salivaire. Il est important à ce stade de mettre le patient en position latérale de sécurité pour libérer les voies aériennes supérieures. Puis le patient présente des signes de réveil, progressif, marqué par une confusion et parfois une agitation. Le sujet ne garde aucun souvenir de la crise et de sa période post-critique. Il existe un décalage temporel entre les premiers signes de réveil objectivé par les témoins oculaires et les premiers souvenirs souvent plus tardifs rapportés par le patient («premier souvenir dans l'ambulance ou au service d'accueil des urgences »). Enfin, la phase post-critique comporte souvent des céphalées, des courbatures, des douleurs en relation avec la morsure latérale de la langue, le traumatisme occasionné par la chute voire avec une luxation d'épaule ou un tassement vertébral survenus au cours de la phase tonique.
b. Crise myoclonique
Ce sont les seules crises généralisées sans trouble de la conscience : elles sont très brèves (< 1 seconde à quelques secondes) et comportent des secousses musculaires très brèves (< 200 ms), isolées ou répétées en courtes salves, en flexion-extension, avec lâchage ou projection de l'objet tenu (signe de la tasse de café) voire chute brutale si elles affectent les membres inférieurs. Elles sont spontanées ou provoquées par des stimulations, en particulier une stimulation lumineuse intermittente. Fréquentes immédiatement après le réveil, elles surviennent en pleine conscience (à distinguer des myoclonies physiologiques survenant à l'endormissement).
c. Absence typique
Les absences typiques comportent une rupture du contact complète, de début et fin bru- tales, avec arrêt de l'activité en cours, fixité voire plafonnement du regard pendant quelques secondes.
L'EEG au cours de l'absence typique se caractérise par une décharge paroxystique généralisée, bilatérale, symétrique et synchrone de pointes-ondes à 3 Hz, de début et fin brusques, de quelques secondes, interrompant une activité de fond normale. Cet aspect de l'EEG critique est pathognomonique des absences typiques.
B. Crises focales


1. Phénoménologie clinique des crises temporales internes
Elles comportent des sensations subjectives végétatives (sensation épigastrique ascendante, sensation de chaleur), émotionnelles (angoisse) et/ou mnésiques (illusion de déjà-vu, état de rêve), un trouble de la conscience qui est inconstant et secondaire, des automatismes oro-alimentaires de mâchonnement, des automatismes gestuels élémentaires à prédominance distale, répétitifs (émiettement, manipulation), une durée prolongée supérieure à une minute ; les généralisations tonicocloniques secondaires sont exceptionnelles sous traitement.
2. Phénoménologie clinique des crises de la région centrale
Elles comportent des paresthésies et/ou des clonies dont l'évolution traduit l'organisation somatotopique du cortex sensitif et moteur primaire, débutant typiquement au niveau de la main avec une progression ascendant le long du membre supérieur puis l'implication de la face (marche bravais-jacksonienne).


III. Classification et diagnostic syndromique
Un syndrome se définit comme une association non fortuite de signes élémentaires cliniques voire paracliniques. L'identification du syndrome épileptique permet en effet d'orienter la recherche étiologique, le choix du traitement antiépileptique, de formuler un pronostic évolutif de l'épilepsie et d'un éventuel handicap psychiatrique ou cognitif associé.
A. Principes de la classification syndromique
Les syndromes sont définis selon :
- l'âge de début des crises;
- le(s) type(s) de crise(s) prédominant(s);
- l'EEG intercritique et critique;
- l'examen neurologique et neuropsychologique intercritique (normal, ou signes de focalisation dans les formes focales, ou troubles du développement dans les encéphalopathies épileptiques) ;
- le pronostic.
Ils sont représentés dans la figure suivante selon trois axes (figure 15.2) : un axe centrifuge correspondant à l'âge de début, un axe vertical correspondant au type de crises (focales ou généralisées) et enfin un axe horizontal étiologique (idiopathique, c'est-à-dire d'origine présumée, ou non idiopathique, c'est-à-dire d'origine structurelle ou métabolique ou infectieuse ou dysimmune).
B. Quelques syndromes à connaître
1. Épilepsie-absence de l'enfant
- Appartient aux épilepsies généralisées d'origine génétique présumée (= idiopathique).
- Âge de début : autour de 6 ans.
- Type de crises prédominant : absences typiques (jusqu'à 100 par jour).
- Autres types de crises associés possibles : crises généralisées tonicocloniques, rares et tardives dans l'évolution, myoclonies.
- Pronostic : pharmacosensible dans 80 % des cas ; possibilité d'arrêter le traitement après la puberté.
- EEG : décharges de pointes-ondes généralisées synchrones à 3 cycles/s, favorisées par l'hyperpnée (figure 15.3).
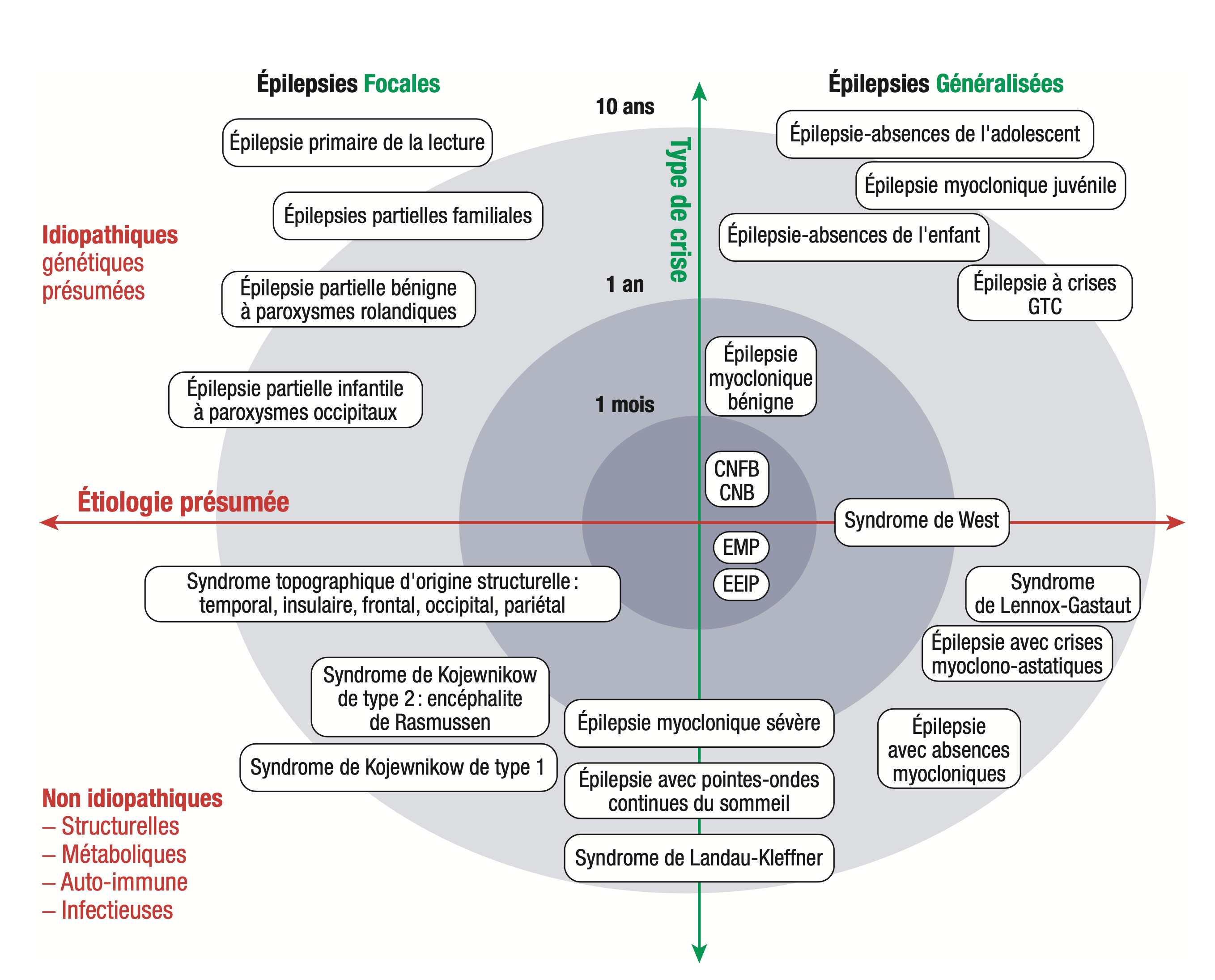
Fig. 15.2. Classification des syndromes épileptiques selon l'étiologie,le typedecrise et l'âge de début.
CNB(F), convulsions néonatales bénignes (familiales); EMP, épilepsies myocloniques progressives; EEIP,
encéphalopathie épileptique infantile précoce.
(Source : CEN, 2019, illustration de Carole Fumat, d'après S. Nguyen.)
2. Épilepsie myoclonique juvénile
- Appartient aux épilepsies généralisées d'origine génétique présumée (= idiopathique).
- Âge de début : adolescence.
- Type de crises prédominant : crises myocloniques matinales, souvent photosensibles.
- Autres types de crises associés possibles : crises généralisées tonicocloniques, absences.
- Pronostic : pharmacosensibles dans 80 % des cas, mais pharmacodépendance à l'âge adulte (il est rarement possible d'arrêter le traitement antiépileptique).
- EEG : bouffées de polypointes-ondes généralisées synchrones, favorisées par la stimulation lumineuse intermittente (figure 15.4).
3. Épilepsie avec crises tonicocloniques du réveil
- Appartient au groupe des épilepsies généralisées d'origine génétique présumée (= idiopathique).
- Âge de début : adolescence, adulte.
- Un seul type de crise : généralisée tonicoclonique, favorisée par le manque de sommeil, l'alcool.
- EEG : pointes-ondes et pointes généralisées.
- Pronostic : pharmacosensible dans 90 % des cas à condition que les règles d'hygiène concernant le sommeil et l'alcool soient bien respectées.
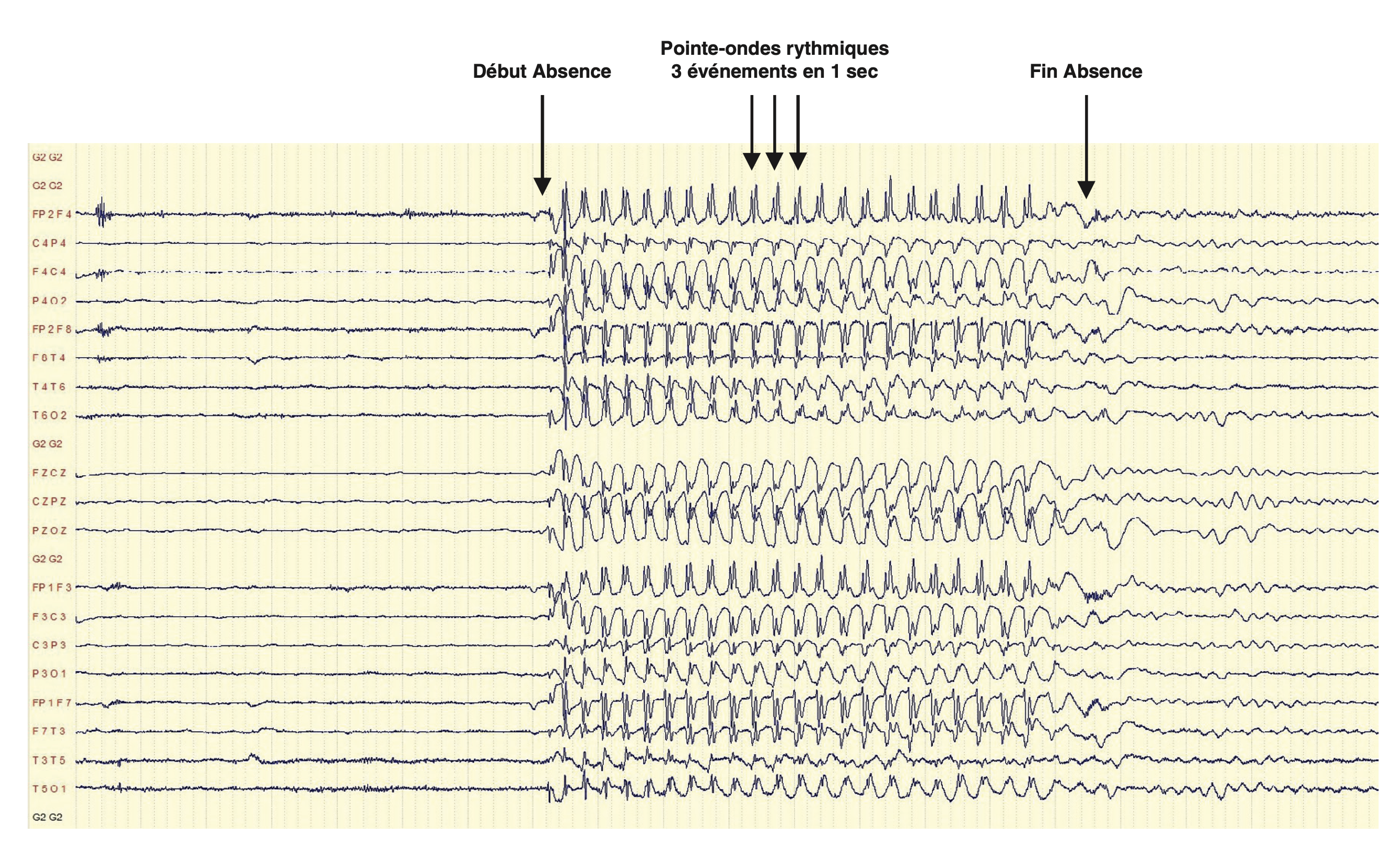
Fig.15.3. Décharge de pointes-ondes généralisées synchrones à 3Hz au cours d'une absence typique.
(Source : CEN, 2019.)
4. Syndrome de West
- Encéphalopathie épileptique liée à l'âge qui appartient aux groupes des épilepsies avec crises généralisées ou avec crises focales ou de début inconnu, et dont les étiologies peuvent également être diverses (génétique présumée, structurelle...).
- Âge de début : 6 mois.
- Un type de crise prédominant, les spasmes en flexion.
- Défini par l'association avec une régression psychomotrice et une hypsarythmie à l'EEG.
- Pronostic variable mais avec un risque élevé de pharmacorésistance et de troubles permanents du développement.
5. Syndromes spéciaux : crise hyperthermique simple et crise hyperthermique compliquée
- Souvent d'origine génétique.
- Elles répondent à des critères diagnostiques très stricts.
- Âge de début après 1 an.
- Crise survenant dans un contexte de fièvre >37,5 °C, comportant des manifestations motrices symétriques, durant moins de 15 minutes, sans déficit post-critique, restant unique pour un épisode fébrile donné.
- Moins de trois épisodes au total.
- Si ces conditions sont remplies, il n'y a pas d'indication d'examen complémentaire, notamment pas d'EEG, et pas d'indication de traitement antiépileptique au long cours. La prévention se fait par un contrôle de l'hyperthermie.
- Lorsque l'un de ces critères n'est pas rempli, on parle de crises hyperthermiques compliquées, qui justifient alors un bilan étiologique et un traitement antiépileptique de fond.
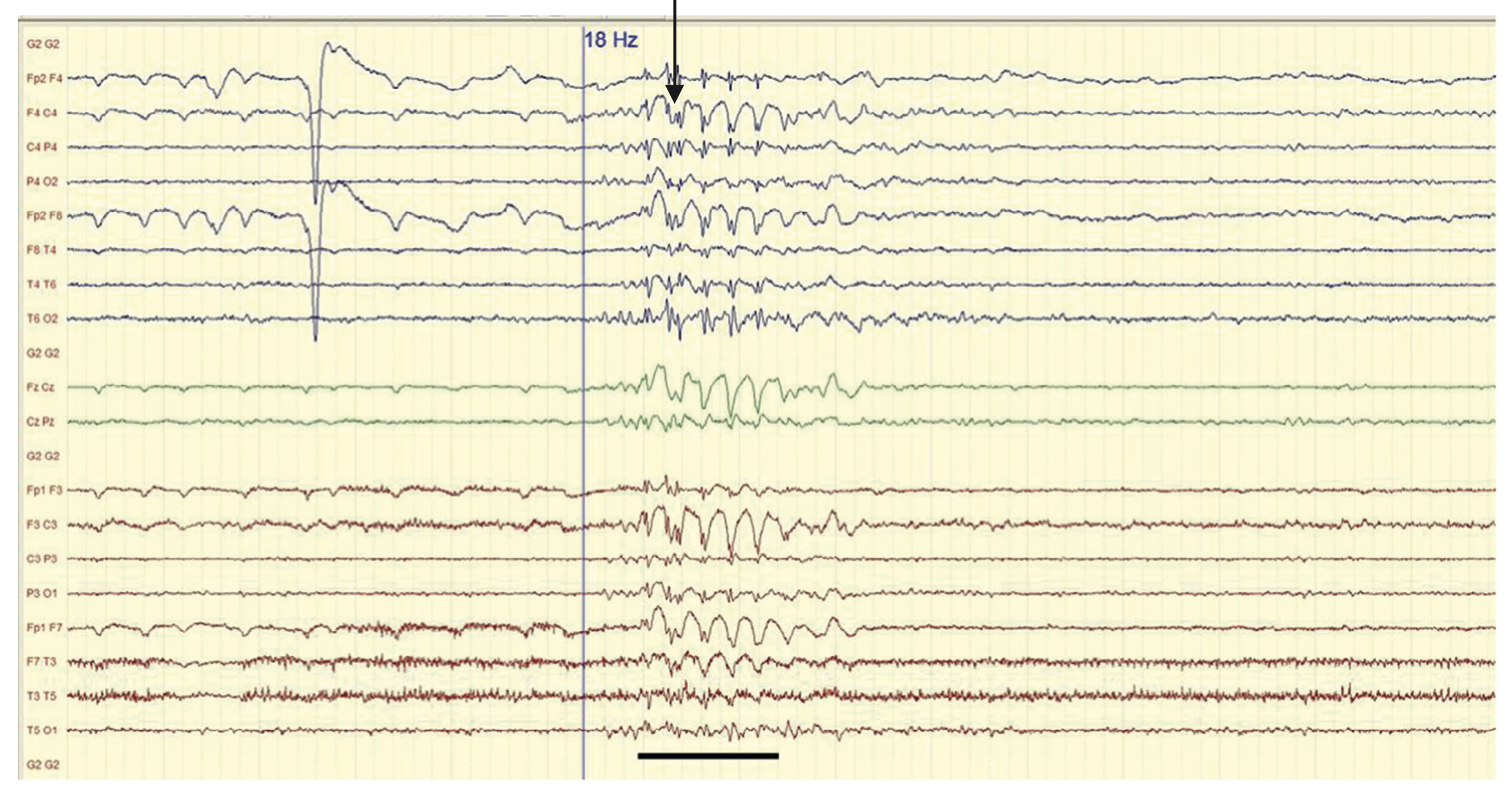
Fig. 15.4. Brève bouffée de polypointes-ondes généralisées synchrones.
(Source : CEN, 2019.)
6. Épilepsie temporale médiale sur sclérose de l'hippocampe
- Antécédents de crises hyperthermiques compliquées.
- Âge de début des premières crises non fébriles : 5 à 15 ans.
- Types de crises prédominants : crises focales comportant des sensations subjectives végétatives (sensation épigastrique ascendante), émotionnelles (angoisse) et/ou mnésique (déjà vu, état de rêve), un trouble de la conscience qui est secondaire, des automatismes de mâchonnement, des automatismes gestuels élémentaires (émiettement, manipulation), une durée prolongée supérieure à 1 minute; crise focale secondairement généralisée exceptionnelle sous traitement.
- Pronostic : mauvais, avec pharmacorésistance fréquente mais accessible alors à un traite- ment chirurgical qui permet la guérison dans 70 % des cas.
- IRM : aspect de sclérose hippocampique unilatérale (figure 15.5).
IV. Diagnostic étiologique
Les crises épileptiques sont des symptômes. Les étapes diagnostiques comportant une description phénoménologique de la (les) crise(s) présentée(s) par le patient, l'identification du type de crise, l'identification du syndrome vont finalement orienter le diagnostic étiologique.
Les causes des épilepsies sont classées dans cinq grandes catégories qui ne sont pas exclusives les unes des autres :
- causes génétiques : 40 % des épilepsies, mais seules quelques-unes sont accessibles à un diagnostic génétique précis ; les autres sont rapportées à une origine génétique présumée sur les données électrocliniques, l'histoire familiale et sont dites «génétiques présumées» (anciennement idiopathiques);
- causes structurelles (lésionnelles) : peuvent être congénitales (malformations corticales telles que les dysplasies corticales, les polymicrogyries, malformations vasculaires telles que les cavernomes) ou acquises (post-traumatique, tumorale, vasculaire) ;
- causes inflammatoires ou dysimmunes (encéphalites auto-immunes);
- causes infectieuses (post-méningitique, post-encéphalitique);
- causes métaboliques : peuvent être secondaires à une cause génétique (comme le syndrome de De Vivo) ou acquise.
Certaines étiologies peuvent parfois être classées dans plusieurs catégories. Par exemple, la sclérose tubéreuse de Bourneville, qui est une malformation cérébrale génétique- ment déterminée, est classée à la fois dans la catégorie structurelle et dans la catégorie génétique.
À de rares exceptions près, il n'y a pas de correspondance stricte entre diagnostic syndromique et génétique : un même génotype peut correspondre à plusieurs phénotypes différents et l'inverse est vrai également.
Lorsque aucune cause n'est suspectée ou identifiée, l'étiologie est dite «inconnue».
La figure 15.6 synthétise le cadre de classification des épilepsies.
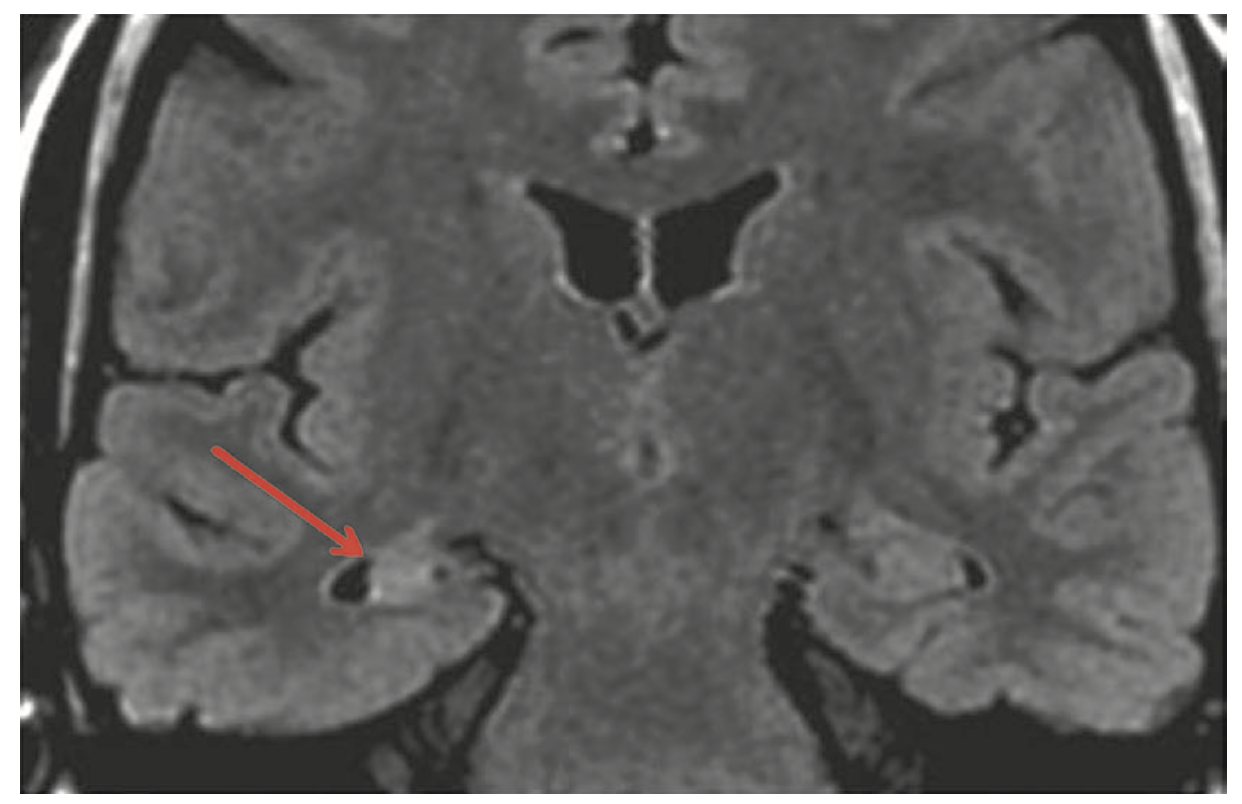
Fig. 15.5. IRM cérébrale en coupe coronale et séquence FLAIR montrant une atrophie associée à une sclérose (hypersignal) de l'hippocampe droit (flèche rouge).
(Source : CEN, 2019.)
V. Démarche diagnostique
Différentes étapes sont nécessaires pour établir le diagnostic (figure 15.7).
A. Étape 1 – Diagnostic positif d'une crise d'épilepsie non symptomatique aiguë
Dans la démarche diagnostique positive d'une épilepsie, la première étape consiste en la confirmation de la nature épileptique du malaise initial et de son origine non provoquée. Est exclue de fait de ce chapitre la prise en charge des crises symptomatiques aiguës (crises sur lésion cérébrale aiguë, troubles métaboliques ou toxiques...). Cette première étape diagnostique est traitée dans le chapitre 30, item 342.  Il ne faut pas confondre avec cette étiologie symptomatique aiguë d'éventuels facteurs précipitants de la crise initiale tels que la privation de sommeil, la stimulation lumineuse intermittente (qui témoigne alors d'une photosensibilité présente dans seulement 5 % des épilepsies).
Il ne faut pas confondre avec cette étiologie symptomatique aiguë d'éventuels facteurs précipitants de la crise initiale tels que la privation de sommeil, la stimulation lumineuse intermittente (qui témoigne alors d'une photosensibilité présente dans seulement 5 % des épilepsies).
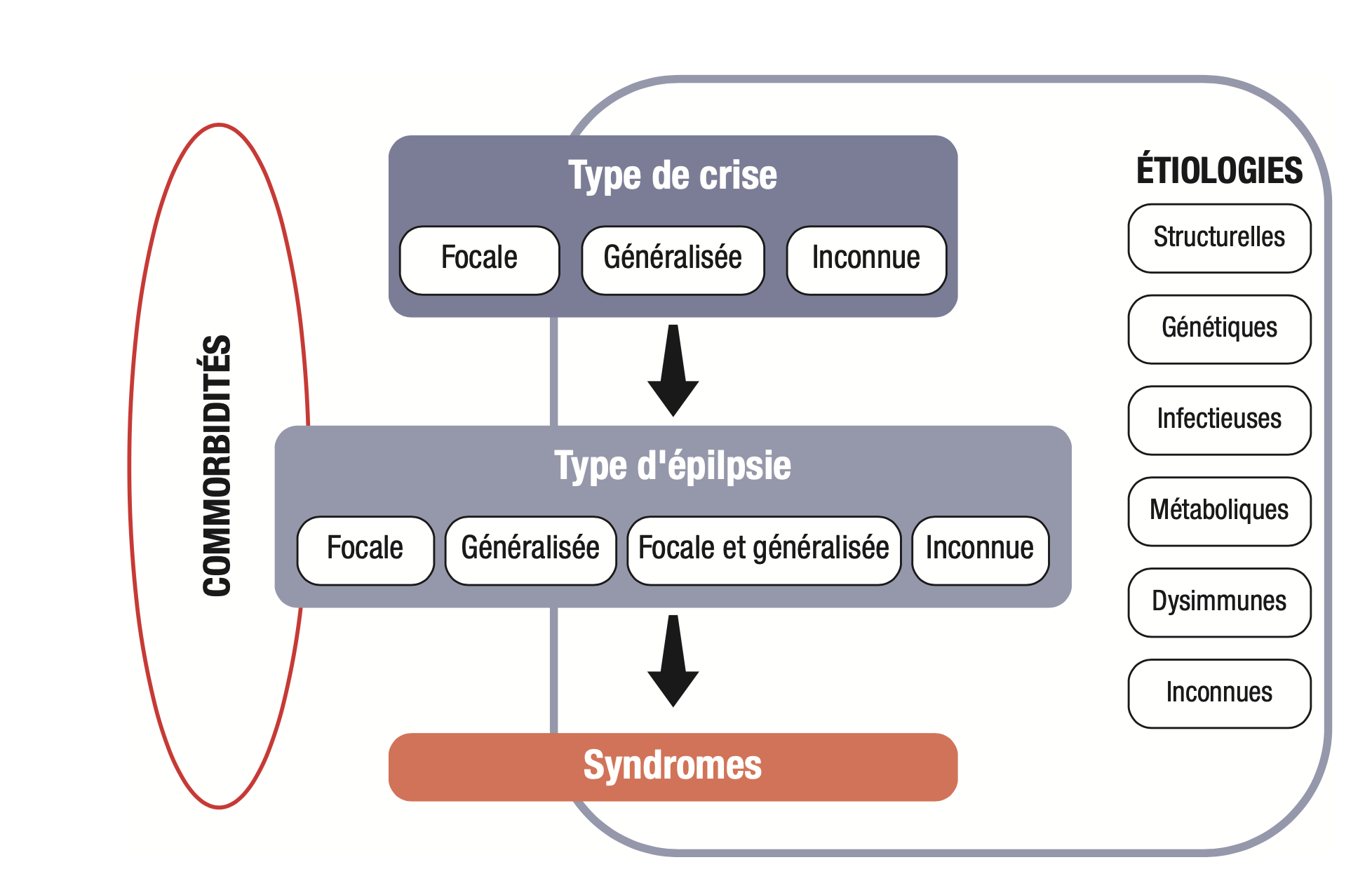
Fig. 15.6. Principes de la classification des épilepsies.
(Source : CEN, 2019, illustration de Carole Fumat.)
B. Étape 2 – Diagnostic différentiel d'une crise d'épilepsie
Les erreurs diagnostiques sont fréquentes à cette étape. Les causes «mimant» une crise d'épilepsie sont fréquentes et parfois graves.
Devant une suspicion de crise généralisée tonicoclonique, principalement deux autres diagnostics peuvent être discutés :
- les syncopes convulsivantes : contexte particulier (effort, miction, toux, douleur...), signes lipothymiques associés, quelques secousses possibles répétées, mais reprise de conscience rapide, absence de confusion post-critique et d'amnésie critique. La perte d'urine et la morsure de langue (pointe classiquement) sont possibles ;
- les crises non épileptiques psychogènes (CNEP) : fréquent contexte psychologique avec traumatismes anciens (sévices corporels, sexuels, psychologiques...), symptomatologie critique polymorphe et très prolongée (mouvement de négation de la tête, balancement du bassin, pleurs, persistance des yeux clos, résistance à l'ouverture des yeux, activité motrice asynchrone et irrégulière...). Parfois les CNEP peuvent être difficiles à différencier au plan sémiologique d'une crise épileptique et seul l'enregistrement simultané vidéo-EEG permet le diagnostic (absence d'activité EEG épileptique durant la CNEP).
Devant une suspicion de crise focale, peuvent être discutés les diagnostics différentiels suivants :
- aura migraineuse : contexte de migraine le plus souvent connu, marche migraineuse plus progressive et plus longue qu'une crise épileptique : sur une période de 5 à 60 minutes, succession plus ou moins complète d'hallucinations visuelles à type de phosphènes puis troubles sensitifs à type de paresthésies latéralisées puis de troubles phasiques (manque du mot, paraphasies...). Les céphalées apparaissent en fin de séquence et peuvent alors persister plusieurs heures;
- accident ischémique transitoire : fréquent contexte de facteurs de risque cardiovasculaire, déficit neurologique transitoire correspondant à un territoire vasculaire avec imagerie normale. La répétition d'un malaise de sémiologie stéréotypée doit remettre en cause ce diagnostic et faire évoquer une crise d'épilepsie ;
- malaise hypoglycémique : contexte de diabète, intoxication à l'insuline...; des phénomènes déficitaires neurologiques jusqu'au coma peuvent être observés durant une hypoglycémie sévère. De manière associée sont constatés des phénomènes hyperadrénergiques : agitation, tremblements, sueurs, tachycardie, hypertension. Le resucrage permet la correction de l'ensemble des troubles neurologiques et confirme le diagnostic.
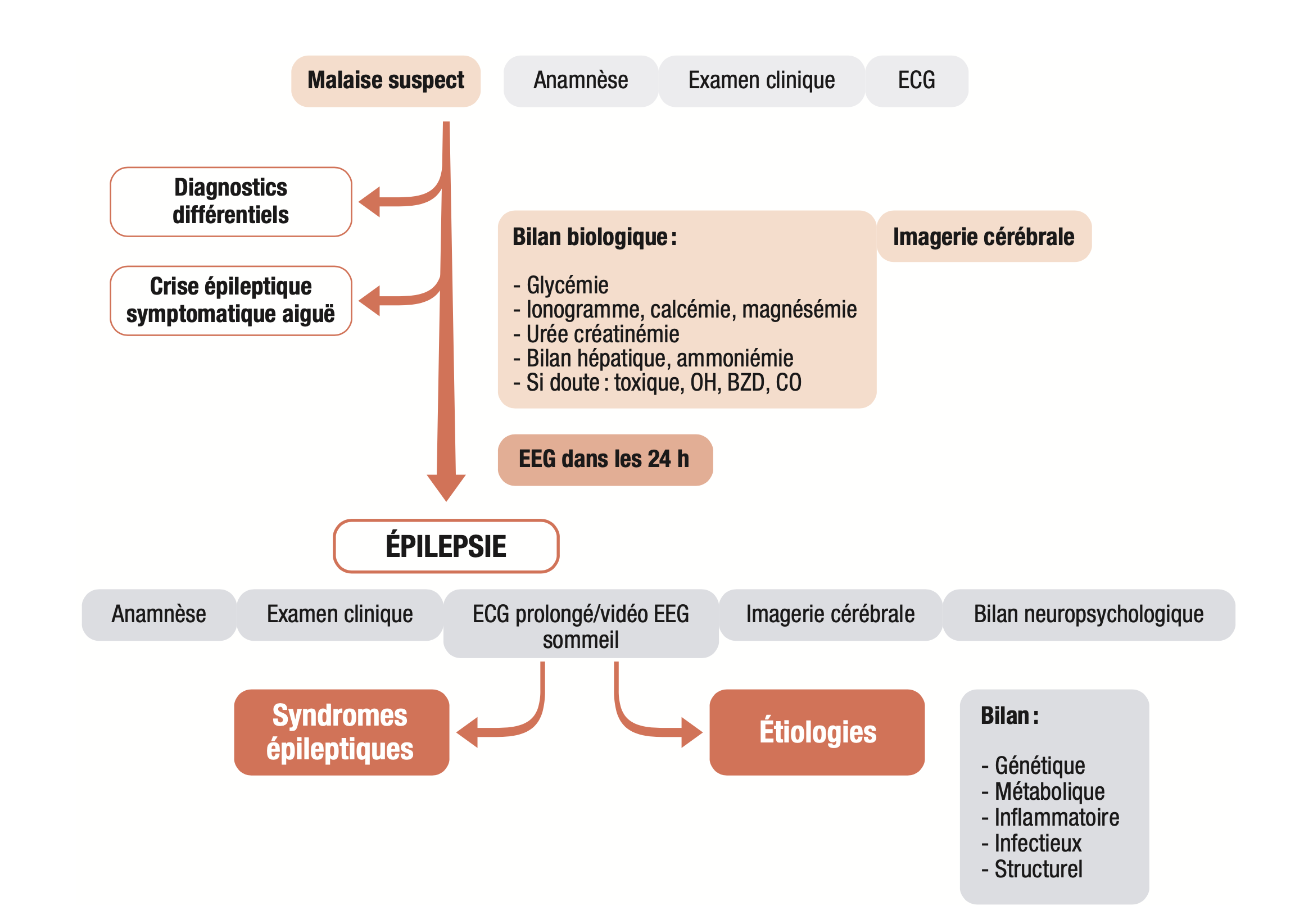
Fig. 15.7. Étapes nécessaires au diagnostic positif d'une épilepsie.
(Source : CEN, 2019, illustration de Carole Fumat.)
C. Étape 3 – Diagnostic positif d'une épilepsie
Cette étape consiste à réunir les arguments en faveur de l'existence d'une prédisposition cérébrale durale à générer des crises (cf « I. Definitions ») associée à la crise épileptique initiale.
Elle va prendre en compte les facteurs de risque associés tels que l'existence d'une lésion cérébrale préexistante à l'imagerie, d'antécédents de retard du développement, d'un examen neurologique anormal, d'anomalies paroxystiques partielles ou généralisées à l'EEG. Un seul de ces éléments associé à une crise épileptique cliniquement avérée suffit à poser le diagnostic de maladie épileptique, le risque de récidive de crise étant élevé (plus de 60 % en 10 ans).
Il convient donc de mener une enquête précise :
- anamnèse : recherche d'événements antérieurs suspects de crises passées inaperçues (par exemple, myoclonies, absences, pertes de connaissance itératives, épisodes de morsure de langue ou de perte d'urines nocturne...);
- examen neurologique intercritique : recherche de signes de focalisation, retard des acquisitions ;
- imagerie cérébrale : la recherche d'une lésion épileptogène en dehors de syndromes d'origine génétique précis repose sur l'IRM encéphalique, qui est systématique. Elle peut être réalisée en différé si un scanner cérébral a déjà été réalisé lors de la prise en charge en aigu de la crise;
- un EEG : recherche des éléments paroxystiques épileptiques. Leur présence permet d'affirmer de façon rétrospective l'origine épileptique d'une crise suspectée cliniquement et constitue également un facteur pronostique de récidive et un critère essentiel pour le diagnostic syndromique. Selon la typologie des crises, l'épilepsie sera définie comme focale, généralisée ou de forme inconnue (cf. supra).
D. Étape 4 – Diagnostic syndromique et étiologique
L'enquête étiologique et l'enquête syndromique s'effectuent de manière conjointe. Elles sont essentielles afin de guider le pronostic et la stratégie thérapeutique : certaines molécules peuvent aggraver des épilepsies de syndrome et/ou de cause spécifique (notamment génétiques et métaboliques).
Le diagnostic étiologique vise à rechercher les causes de l'épilepsie. Le diagnostic étiologique est guidé par le diagnostic syndromique. Un syndrome épileptique regroupe les épilepsies présentant un profil clinique, paraclinique, pronostique et de réponse thérapeutique identique mais pouvant être associées à des étiologies distinctes.
Les diagnostics étiologique et syndromique reposent avant tout sur :
- une anamnèse précise du patient et de son entourage afin de documenter les antécédents personnels et familiaux, l'histoire de la maladie épileptique :
- personnels :
- antécédents : souffrance fœtale aiguë à la naissance, crise convulsive fébrile dans l'enfance, notion de méningite ou d'encéphalite, traumatisme crânien sévère, retard des acquisitions psychomotrices, malformations congénitales, difficultés scolaires,
- pathologies associées : surdité, pathologies auto-immunes, troubles psychiatriques...,
- histoire de l'épilepsie : âge au début des crises, évolution du type, de la fréquence et sémiologie des crises, essais thérapeutiques, évolution cognitive et psychiatrique parallèlement à celle de l'épilepsie,
- familiaux : épilepsie, retard des acquisitions, malformations congénitales, pathologies neurologiques, pathologies auto-immunes...;
- personnels :
- un examen neurologique et général complet : syndrome cérébelleux, syndrome pyra- midal, déficit moteur focal, champ visuel, recherche d'une atteinte cutanée, trouble du langage, surdité...;
- une IRM cérébrale avec recherche d'une lésion épileptogène (par exemple, séquelles vasculaires ou traumatiques, malformation de développement cortical, sclérose hippocampique, cavernome...);
- un enregistrement EEG, si besoin complété par un enregistrement vidéo-EEG prolongé avec sommeil : identification des éléments paroxystiques intercritiques, parfois des crises électrocliniques au repos ou lors des manœuvres d'activation (hyperpnée, stimulation lumineuse intermittente). L'enregistrement vidéo-EEG, prolongé afin d'obtenir des enregistrements durant le sommeil, est plus sensible à la détection de paroxysmes EEG ;
- un bilan neuropsychologique : recherche d'une atteinte cognitive spécifique associée à la symptomatologie des crises, comorbidité dépendant du diagnostic étiologique et syndromique.
La synthèse diagnostique initiale doit être réalisée par un spécialiste neurologue ou neuropédiatre.

VI. Diagnostic de gravité : une urgence vitale, l'état de mal épileptique généralisé tonicoclonique

D'un point de vue opérationnel, il existe non pas un mais des états de mal épileptique qui se définissent comme des crises anormalement prolongées qui peuvent mettre en jeu le pronostic vital ou fonctionnel du patient. La durée des crises étant variable selon le type de crise, la durée au-delà de laquelle le diagnostic d'état de mal épileptique est posé dépend du type de crise. En ce qui concerne les crises tonicocloniques généralisées, un diagnostic d'état de mal épileptique est posé si les phases toniques et cloniques réunies durent plus de 5 minutes.

Pour les autres formes d'état de mal (non convulsives, d'expression confusionnelle), la durée au-delà de laquelle le diagnostic peut être posé est variable, entre 10 et 30 minutes. De même, la mise en jeu du pronostic vital et fonctionnel est retardée et le diagnostic plus difficile : dans ces cas, l'urgence est de faire un diagnostic précis (en évitant le surdiagnostic). Celui-ci repose sur une analyse clinique fine corrélée à la réalisation d'un EEG en urgence.
VII. Principes thérapeutiques
A. Conduite à tenir en présence d'une crise généralisée tonicoclonique
- Noter l'heure de début (afin de pouvoir en déterminer la durée qui conditionne le diagnostic de gravité de l'état de mal épileptique).
- Libérer les voies aériennes supérieures (ne pas introduire les doigts ou tout objet).
- Position latérale de sécurité (participe à libérer les voies aériennes supérieures, prévient une pneumopathie d'inhalation en cas de vomissements).
- En préhospitalier, s'il s'agit d'une crise habituelle chez un patient épileptique connu et traité, pas d'hospitalisation systématique. S'il s'agit d'une première crise, ou d'une crise inhabituelle, ou si la durée est supérieure à 5 minutes, appel du 15 pour prise en charge au service des accueils des urgences.
- Au SAU, pas de traitement antiépileptique systématique en l'absence d'état de mal épileptique.
6 Gastaut H., Poire R., Roger J., Lob H., « Les états de mal généralisés tonico-cloniques et les états de mal épileptiques », in H. Gastaut, J. Roger, H. Lob (éds.), Les États de mal épileptiques, Paris, Masson, 1967, p. 11-43.
B. Prise en soin médicamenteuse : indication d'un traitement antiépileptique de fond : est-il nécessaire d'introduire un traitement antiépileptique de fond?
Il s'agit d'une question fondamentale. Un traitement antiépileptique ne doit pas être commencé de manière systématique devant toute première crise d'épilepsie.
Seuls les patients présentant un risque de récidive élevé de crises à long terme doivent être traités. En l'absence de tels critères, il n'existe aucune indication de traitement antiépileptique même temporaire (par exemple, benzodiazépines).
Les patients pour lesquels une épilepsie a été diagnostiquée sont par définition à haut risque de récidive. L'introduction de ce traitement doit cependant s'effectuer après une information claire et avec l'accord du patient. Ce dernier doit en comprendre les risques et bénéfices attendus.
C. Prise en charge médicamenteuse de l'état de mal épileptique
L'état de mal épileptique est la seule indication d'un traitement antiépileptique urgent de la crise. Seul l'état de mal épileptique convulsif (généralisé tonicoclonique) a fait l'objet de recommandations d'experts (Société de réanimation de langue française et Société française de médecin d'urgence), actualisées en 2018.
Les principes de la prise en charge médicamenteuse sont résumés dans la figure 15.8.
1. En cas d'intervention avant 30 minutes
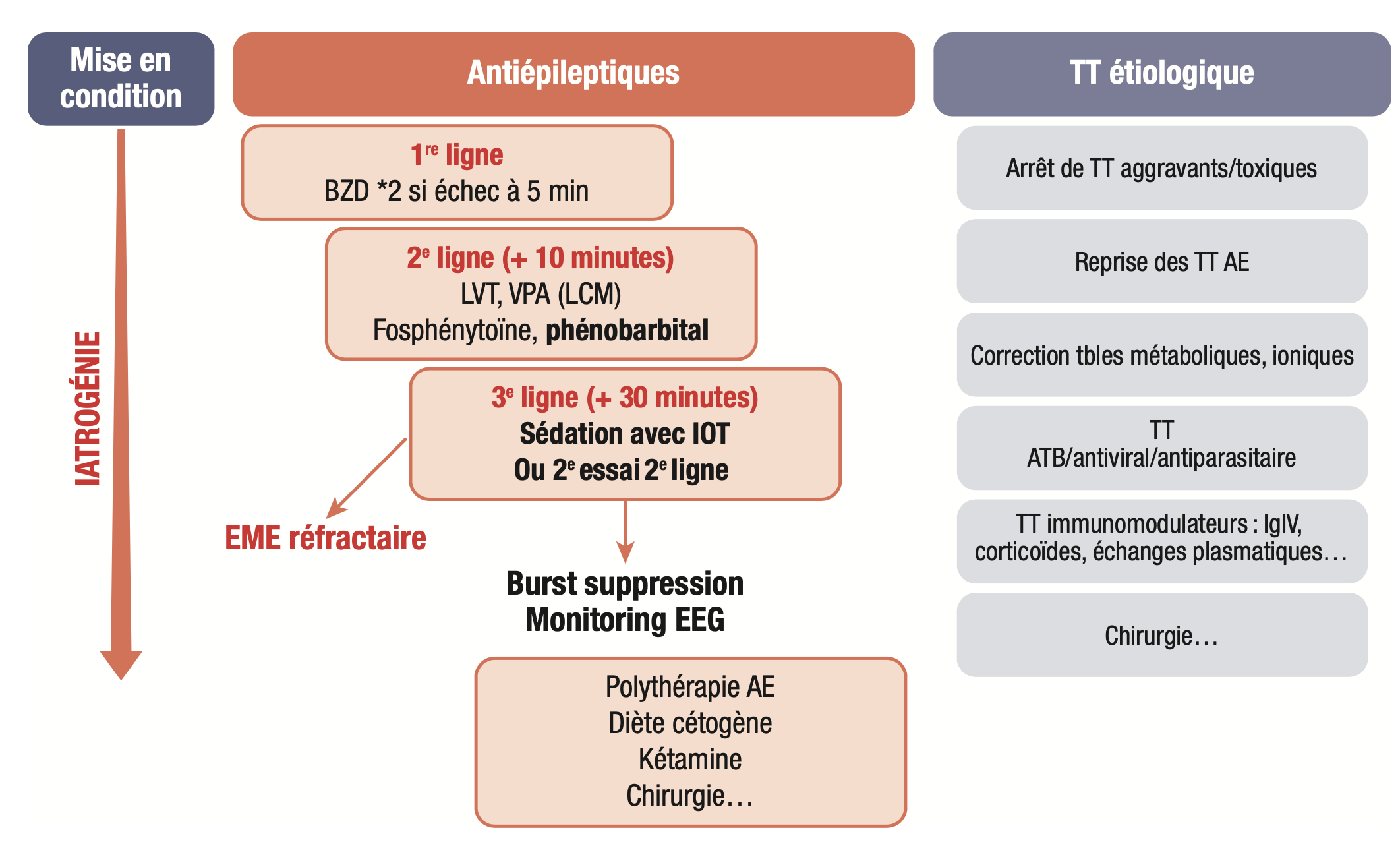
Fig. 15.8. Prise en charge médicamenteuse de l'état de mal épileptique.
AE, antiépileptiques ; EME, état de mal épileptique ; LVT, lévétiracétam ; VPA, acide valproïque ; LCM, lacosamide ; IOT, intubation orotrachéale ; TT, traitement.
(Source : CEN, 2019, illustration de Carole Fumat.)
1. Première ligne de traitement : benzodiazépine en intraveineuse lente (clonazépam 1 mg), répété une fois si échec au bout de 5 minutes.
2. Deuxième ligne de traitement : en cas d'échec 5 minutes après la deuxième injection de benzodiazépine, antiépileptique d'action prolongée en IV à la seringue électrique : fosphénitoïne (Prodilantin®) 20 mg/kg d'équivalent phénitoïne ou phénobarbital (Gardénal®) 15 mg/kg, ou lévétiracétam 30–60 mg/kg < 4 g, acide valproïque 40 mg/kg et < 3 g, en IV, lacosamide 200 mg de dose de charge.
3. Troisième ligne de traitement : si persistance de l'état de mal 30 minutes après l'administration de la deuxième ligne, sédation (propofol ou midazolam ou thiopental) avec intubation orotrachéale. Cette situation correspond à l'état de mal épileptique réfractaire.
Toutefois, si l'intubation orotrachéale apparaît déraisonnable (limitation thérapeutique) ou s'il s'agit d'un patient ayant une épilepsie connue et que l'état de mal épileptique évolue depuis moins de 60 minutes, ou chez l'enfant, il est possible de retarder le coma thérapeutique et d'avoir recours à un autre antiépileptique de deuxième ligne en cas d'état de mal réfractaire.
2. En cas d'intervention après 30 minutes
Benzodiazépine et antiépileptique d'action prolongée en IV de deuxième ligne.
3. Dans tous les cas
- Mise en condition du patient (libération des voies aériennes supérieures, oxygénothérapie, voie veineuse périphérique, surveillance en soins continus voire en réanimation médicale (SaO2, FC, FR, TA, glycémie, ionogramme sanguin, pH).
- Traitement étiologique.
- Mise en place rapide d'un traitement antiépileptique de relais de la dose de charge.
D. Prise en charge non médicamenteuse chronique : informations à donner aux patients
De manière toute aussi importante que le traitement médicamenteux, la prise en soin d'un patient épileptique repose sur une éducation complète. L'observance, la tolérance et l'efficacité thérapeutique dépendent principalement de la qualité de cette information.
Plusieurs points devront être abordés au cours des premières consultations (cf. encadré ci-dessous).