Une femme de 55 ans présentait depuis deux 9 ans des troubles de l'équilibre avec sensation de tangage, des chutes en rétropulsion et une dysarthrie hypophonique. Un an après le début des symptômes apparurent des troubles de déglutition prédominant sur les liquides et une détérioration cognitive marquée par un score de 126/144 à l'échelle de Mattis. Elle présentait comme principaux antécédents une coronaropathie et plusieurs interventions chirurgicales (chirurgie de l'uretère droit, cure de hernie inguinale et ablation de kystes mammaires). Lors de la première consultation, son traitement comportait 100 mg de piribédil et 250 mg de lévodopa associée à la bensérazide. L'examen neurologique objectivait un syndrome cérébelleux cinétique et statique, un syndrome frontal, une instabilité posturale avec rétropulsion, des réflexes ostéotendineux vifs et un ralentissement des saccades oculaires. Il n'y avait pas de syndrome parkinsonien segmentaire, ni de dysautonomie.
Biologiquement, il n'y avait pas de carence en vitamines du groupe B (B1, B6, B9 et B12), ni en vitamine E, la recherche d'anticorps anti- GAD était négative. Le bilan métabolique, comprenant la chromatographie des acides aminés et l'étude pré- et post-prandiale sur 24 h de l'ammoniémie et du rapport lactate/ pyruvate ne montrait pas d'anomalies. L'EEG, l'IRM cérébrale et la ponction lombaire ne donnaient pas d'arguments en faveur d'une maladie de Creutzfeldt-Jacob.
Environ un an plus tard, apparurent de discrets mouvements choréiques et un syndrome extrapyramidal segmentaire. L'étude en biologie moléculaire des gènes IT15, ADRLP et SCA17 était négative. Dans les mois qui suivirent, s'installa progressivement un rétrocolis, une ophtalmoparésie franche de la verticalité du regard. À l'examen, il existait une nette aggravation des troubles posturaux et du syndrome frontal avec apparition d'un signe de l'applaudissement franc.
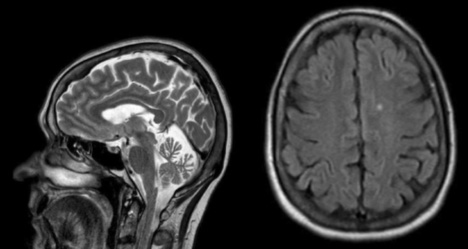
- Comment analyser-vous l'IRM ?
- Quel diagnostic évoquez-vous ?
- Quel examen complémentaire demandez-vous pour l'affirmer ?
G Grolez, A Dejardin, D Devos, L Defebvre, C Moreau (Lille).
- L’ IRM cérébrale, à gauche en séquence T2 sagittale montre une atrophie vermienne. À droite, en séquence FLAIR axiale elle montre un hypersignal non significatif de substance blanche.
- L ‘association d’une ataxie cérébelleuse, d’une ophtalmoplégie supranucléaire, d’une dysarthrie, d’une dysphagie, de troubles cognitifs, et de mouvements anormaux conduit à évoquer le diagnostic de maladie de Niemann Pick de type C
- Une mise en culture de fibroblastes cutanés pour réaliser un test à la filipine et le séquençage des gènes NPC1 et NPC2.
Références :
Godeiro-Júnior C, Inaoka RJ, Barbosa MR, Silva MR, Aguiar Pde C, Barsottini O. Mutations in NPC1 in two Brazilian patients with Niemann-Pick disease type C and progressive supranuclear palsy-like presentation. Mov Disord. 2006;21(12):2270–2.
Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16.
Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F; NP-C Guidelines Working Group. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab. 2012;106(3):330–44.
Wijburg FA, Sedel F, Pineda M, Hendriksz CJ, Fahey M, Walterfang M, et al. Development of a suspicion index to aid diagnosis of Niemann-Pick disease type C. Neurology 2012;78 (20):1560–7 [doi: 10.1212/WNL.0b013e3182563b82. Epub 2012 Apr 18].
La maladie de Niemann Pick de type C est une pathologie du trafic intracellulaire du cholestérol de transmission autosomique récessive. Son expression est extrêmement variable en fonction de l'âge de début des symptômes. Un score prédictif a été réalisé pour aider au diagnostic (Wijburg et al). Le bilan diagnostique comporte une mise en culture de fibroblastes par biopsie cutanée pour réaliser un test à la filipine et le séquençage du gène NPC1 et NPC2. L'interprétation des tests se fait de manière combinée.
A l'âge adulte, le tableau clinique peut associer par ordre de fréquence une ataxie cérébelleuse, une ophtalmoplégie supranucléaire, une dysarthrie, des troubles cognitifs, des mouvements anormaux (syndrome parkinsonien, dystonie, chorée), des troubles psychiatriques et une dysphagie. La splénomégalie est beaucoup moins fréquente à l'âge adulte, l'épilepsie est rare.
L'âge de début est assez tardif chez cette patiente. La biopsie cutanée et la biologie moléculaire confirmèrent une maladie de Niemann Pick de type C avec une mutation hétérozygote c.1843C>T, c3019C>G.