Le concept de protéinopathies
Charlotte Dubucs , Monique Courtade-Saidi , Thibaud Lebouvier et Mathieu
Les cellules les plus nombreuses au sein du système nerveux central sont les neurones, les astrocytes et les oligodendrocytes (cf. chapitre 2). Les neurones sont spécialisés dans la réception, l'intégration et l'émission de signaux nerveux, la transmission de l'information à d'autres neurones étant en grande majorité médiée par des neurotransmetteurs, au niveau synaptique. Les astrocytes jouent de nombreux rôles notamment mécanique de soutien, métabolique, et assurent le développement et le maintien de la barrière hématoencéphalique. Les oligodendrocytes jouent un rôle essentiel de myélinisation des fibres nerveuses de la substance blanche.
S'il existe un renouvellement des astrocytes et des oligodendrocytes, les neurones sont des cellules différenciées qui ne se multiplient plus. Une régénération peut avoir lieu à partir de cellules souches neurales mais elle est difficile à estimer chez l'Homme et très certainement insuffisante pour assurer un remplacement neuronal dans les maladies neurodégénératives (MND). Aussi leur intégrité et le maintien de leur environnement sont capitaux.
La plupart des MND se traduisent par une accumulation et une agrégation de protéines intra ou extracellulaires dans le parenchyme cérébral, souvent en raison de l'adoption par ces dernières d'une conformation anormale. Les agrégats protéiques intracellulaires sont rapportés sous le nom d'inclusions, cytoplasmiques ou nucléaires selon leur topographie, alors que les agrégats extracellulaires sont désignés sous le terme de dépôts. Ces « protéinopathies » occupent une place centrale dans la neurotoxicité et la propagation de la pathologie de certaines MND (tableau 28.1).
Tableau 28.1
Classification moléculaire des maladies neurodégénératives (liste non exhaustive).
| Topographie de l'agrégat | Nature de la protéine ou du peptide | Caractéristiques de l'agrégat | Nom de la maladie |
|---|---|---|---|
| Extracellulaire (dépôt) Maladie d'Alzheimer |
Peptide β-amyloïde Aβ, fragment de la protéine APP (gène APP) : amyloïdopathies | Agrégats périvasculaires à prédominance de Aβ40 (peptide β-amyloïde à 40 acides aminés) | Angiopathie amyloïde cérébrale |
| Plaques amyloïdes (autrefois appelées plaques séniles) parenchymateuses, à prédominance de Aβ42 (peptide β-amyloïde à 42 acides aminés) | |||
| Trisomie 21 (syndrome de Down ) | |||
| Protéine prion PrP (gène PRNP) : maladies à prions | Plaques amyloïdes constituées de PrP | Maladie de Creutzfeldt-Jakob et autres maladies à prion | |
| Intracellulaire (inclusion) Dégénérescence corticobasale Paralysie supranucléaire progressive Sclérose latérale amyotrophique (SLA) Dégénérescences lobaires frontotemporales |
Protéine TAU (gène MAPT) : taupathies | Dégénérescences neurofibrillaires formées d'inclusions de protéine tau à 3 et 4 domaines de fixation aux microtubules (TAU 3R) | Maladie d'Alzheimer |
| Inclusions neuronales et gliales de protéine tau à 4 domaines de fixation aux microtubules (TAU 4R) | |||
| Inclusions neuronales de protéine tau à 3 domaines de fixation aux microtubules | Maladie de Pick (sous-type de dégénérescence lobaire frontotemporale ) | ||
| Protéine TDP-43 (gène TARDP) : tardopathies | 4 types d'inclusions neuronales et gliales décrits | ||
| Alphasynucléine (gène SCNA) : synucléinopathies | Corps de Lewy (inclusions à prédominance neuronale) | ||
| Maladie de Parkinson | |||
| Maladie à corps de Lewy diffus (démence à corps de Lewy) | |||
| Inclusions à prédominance oligodendrocytaire | Atrophie multisystème | ||
| Protéines porteuses d'expansions de polyglutamine (expansion de triplets CAG) | Protéine Huntingtine (gène IT15) | Maladie de Huntington | |
| Ataxines 1 et 3 | |||
| Ataxie spinocérébelleuse de type 1 | |||
| Ataxie spinocérébelleuse de type 3 (maladie de Machado-Joseph) |
Formation des agrégats
La physiopathologie des protéinopathies fait intervenir la formation des premiers oligomères, qui sont les agrégats les plus toxiques pour la cellule. La formation des agrégats survient dès lors qu'une protéine normale adopte une conformation (encadré 28.1) anormale qui la rend prompte à l'agrégation. Toutes les protéines n'ont pas cette tendance agrégative ; il s'agit néanmoins d'une propriété partagée par les peptides et protéines impliqués dans des MND, liée à leur caractère hydrophobe ou à la présence de séquences répétées. Dans des circonstances particulières, ces protéines mal conformées s'assemblent en oligomères solubles, en protofibrilles, puis en fibrilles insolubles qui constituent les agrégats visibles au microscope (cf. figure 28.1). Les cellules sont naturellement dotées d'un système de « contrôle-qualité » assuré par des protéines chaperons qui se lient aux protéines amorphes nouvellement formées à leur sortie du ribosome et assurent qu'elles adoptent la bonne conformation. Les protéines mal conformées sont alors dirigées vers deux voies possibles de dégradation : celle du système ubiquitine-protéasome (encadré 28.1) ou celle des lysosomes (organelle responsable de l'autophagie, encadré 28.1). La première étape du processus dégénératif voit donc la saturation de ces systèmes de contrôle-qualité résultant du vieillissement cellulaire, d'un stress métabolique ou de facteurs génétiques ; les facteurs génétiques peuvent intervenir en accroissant la production ou l'agrégabilité des protéines ou en altérant la fonction des protéines du système ubiquitine-protéasome ou de la voie lysosomale.
Encadré 28.1 Définitions
Biomarqueur
Caractéristique mesurable liée à un processus biologique ou pathologique, qui peut être utilisée à visée diagnostique, pronostique ou théragnostique, c'est-à-dire pour évaluer l'effet d'un traitement. Dans l'acception traditionnelle du terme, les biomarqueurs sont des marqueurs biologiques (ex : biomarqueurs de la maladie d'Alzheimer dans le liquide cérébrospinal) ; par extension, les marqueurs d'imagerie (ex : dénervation dopaminergique en scintigraphie au ioflupane) sont assimilés à des biomarqueurs.
Amyloïde
Ne désigne pas une protéine en particulier mais une conformation secondaire particulière adoptée par des protéines très diverses, ne possédant aucune homologie de séquence. Ces protéines sont susceptibles, dans certaines conditions, de polymériser sous forme d'agrégats fibrillaires dont l'unité est une protofibrille. Chaque protofibrille est constituée d'un assemblage « en pile d'assiettes » de feuillets β de disposition parallèle ou antiparallèle, nommé structure cross-β. La ou les portions amyloïdogénique(s) de la protéine participent à la formation de ces feuillets β. Les dépôts amyloïdes possèdent une forte affinité pour le rouge Congo et la thioflavine. La substance amyloïde est biréfringente en lumière polarisée (deux rayons lumineux sont réfléchis).
Conformation
Les protéines possèdent une structure primaire, secondaire et tertiaire. La structure primaire correspond à sa séquence d'acides aminés. La structure secondaire décrit le repliement local de la chaîne principale, par exemple sous forme d'hélice α ou de feuillet β. La structure tertiaire est la structure tridimensionnelle de la protéine, le repliement de la chaîne polypeptidique dans l'espace. Structures secondaire et tertiaire correspondent à la conformation de la protéine.
Système ubiquitine-protéasome
Système multienzymatique sophistiqué assurant la protéolyse intracellulaire. Le substrat est d'abord « marqué » par conjugaison covalente de chaînes d'ubiquitine (Ub). L'ubiquitine se retrouve donc associée à la majorité des agrégats quelle que soit leur nature, et les anticorps antiubiquitine peuvent être utilisés en histologie pour mettre en évidence les agrégats. Les molécules polyubiquitinylées sont ensuite reconnues et dégradées par un complexe protéolytique de 2 000 kDa, le protéasome 26S.
Autophagie
Processus cellulaire qui permet la dégradation par le lysosome de constituants cytoplasmiques tels que des protéines ou des organites. Il permet la production par la cellule de ressources énergétiques dans des conditions de stress. Il est également essentiel à la mort cellulaire programmée (apoptose).
Espèces réactives d'oxygène
Sous-produits toxiques pro-oxydants du métabolisme normal de l'oxygène, produits lors de la réduction tétravalente de l'oxygène en eau dans la membrane interne des mitochondries.
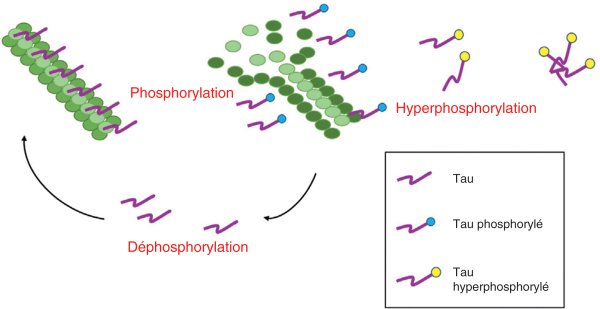
Figure 28.1
Phosphorylation de la protéine tau et son accumulation en agrégats.
La protéine déphosphorylée se lie aux microtubules et les stabilise, la phosphorylation détache tau des microtubules. Une hyperphosphorylation se traduit par des agrégats que l'on retrouve dans des pathologies neurodégénératives.
© Martin CB, Preedy VR. Genetics, neurology, behavior, and diet in Parkinson's disease. The Neuroscience of Parkinson's, Volume 2. Cambridge : Academic Press ; 2020.
Principales protéines impliquées dans les maladies neurodégénératives
Protéine tau
Au niveau des neurones, les microtubules servent de support aux transports axonaux en association avec des protéines associées aux microtubules, assurant par exemple le déplacement de vésicules le long de l'axone. Comme les microtubules sont spontanément des structures dynamiques en perpétuel remaniement (polymérisation et dépolymérisation), leur stabilité est assurée par des protéines qui leurs sont associées.
Au niveau de l'axone, la protéine tau stabilise les microtubules. Cette dernière n'est pas observée au niveau des dendrites. Elle se lie aux microtubules dans sa forme non phosphorylée, elle se détache des microtubules dans sa forme phosphorylée, permettant ainsi leur dépolymérisation. Une hyperphosphorylation de la protéine tau, sur des sites habituellement non phosphorylés dans des conditions physiologiques, s'observe dans la maladie d'Alzheimer (MA). Elle se traduit par son inactivation, l'empêchant de jouer son rôle de stabilisation des microtubules (figure 28.1). Ainsi, les transferts axonaux indispensables au fonctionnement et à la survie du neurone sont inactivés et contribuent au dysfonctionnement et à la mort neuronale. De plus, la protéine tau hyperphosphorylée, insoluble, s'agrège sous forme d'enchevêtrements neurofibrillaires contribuant également à la mort neuronale. Des formes oligomériques pourraient être sécrétées et transmettre la pathologie de cellule à cellule. D'autres MND, caractérisées par des inclusions neurales et gliales de tau, font partie des tauopathies (cf. tableau 33.4).
Peptide β-amyloïde
Il s'agit d'un fragment de protéine transmembranaire (la protéine précurseur de l'amyloïde ou APP) qui joue plusieurs rôles, notamment dans le fonctionnement synaptique des neurones. Un fragment soluble de l'APP est sécrété dans le milieu extracellulaire après un clivage au niveau transmembranaire par une sécrétase α, libérant ainsi un peptide soluble. D'autres sécrétases, dites β et γ, peuvent cliver également l'APP sur des sites différents, aboutissant à la formation d'un peptide hydrophobe appelé β-amyloïde. Le peptide β-amyloïde a une propension à s'agencer en feuillets β insolubles (adoptant une conformation amyloïde au sens physicochimique du terme). Dans le contexte de la MA, ces agrégats s'agglutinent pour former des plaques séniles (ou amyloïdes) extracellulaires. Des oligomères solubles peuvent également diffuser au sein du cerveau, amplifiant les dommages.
On considère généralement que l'amyloïdopathie cérébrale est le pilier principal et l'initiateur de la cascade pathologique qui caractérise la MA, notamment pour des raisons liées à la génétique : les très rares formes autosomiques dominantes de MA sont liées à des gènes impliqués dans la formation du peptide β-amyloïde tels que ceux codant pour l'APP, les présénilines 1 (PSEN-1) et 2 (PSEN-2). L'allèle ε4 de l'apolipoprotéine E (ApoE) – reconnu comme un facteur de risque dans les formes sporadiques – jouerait également un rôle dans la formation des plaques amyloïdes.
α-synucléine
Il s'agit d'une protéine impliquée dans la distribution des lipides de la bicouche membranaire et dans la fusion de vésicules membranaires avec les terminaisons présynaptiques lors de la neurotransmission. La conformation de la protéine est très instable, allant de monomères solubles à des agencements complexes où la protéine est repliée. Comme les autres protéines impliquées dans la neurodégénérescence, l'α-synucléine a une propension à adopter une conformation amyloïde en s'assemblant avec d'autres monomères et à former des agrégats (figure 28.2). Ces agrégats sont souvent rattachés à un feuillet membranaire. Lors de la mort neuronale, ces agrégats peuvent diffuser, être recaptés et provoquer de nouveaux agrégats dans d'autres cellules.
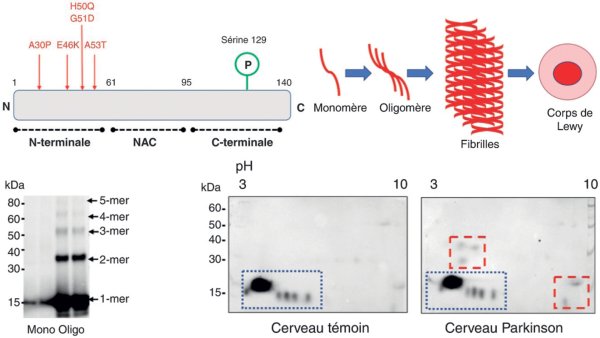
Figure 28.2
Physiologie et physiopathologie de l'alphasynucléine.
Structure et principales régions de l'α-synucléine. L'α-synucléine est divisée en trois domaines : la partie N-terminale est impliquée dans sa liaison aux membranes cellulaires et contient les mutations connues responsables de formes autosomiques dominantes de la maladie, son domaine central est impliqué dans l'agrégation de la protéine, alors que l'extrémité carboxyterminale contient les principaux sites de phosphorylation, en particulier la sérine 129. NAC : non amyloid component.
© SOFMA, Defebre L, Vérin M. La maladie de Parkinson, 4e éd. Paris : Elsevier Masson ; 2020.
Selon le siège intracellulaire et la conformation des agrégats, on peut distinguer plusieurs maladies neurodégénératives en lien avec l'α-synucléine, dites synucléinopathies, dont la plus fréquente est la maladie de Parkinson (cf. tableau 28.1).
Protéine TDP-43
De découverte plus récente, la protéine TDP-43 (Transactive response desoxyribonucleic acid-binding protein), codée par le gène TARDP et associée à l'ARN messager au sein du noyau cellulaire, est impliquée dans différentes MND (cf. tableau 33.4).
Cas particulier de la protéine prion
La protéine du prion cellulaire (PrP) est une glycoprotéine monomérique ancrée à la membrane plasmique qui joue différents rôles tels que dans l'homéostasie du cuivre, le stress oxydatif ou la transduction de signaux cellulaires. La PrP est riche en hélices α, soluble et sensible aux protéases. Le prion (proteinaceous infectious particle) est la forme anormale de la PrP. C'est en acquérant une conformation anormale en feuillets β (protéine prion scrapie, PrPsc) qu'elle montre une résistance accrue aux protéases, elle devient insoluble et agrégable. Elle a ainsi la capacité de progresser d'une cellule à l'autre, d'un organe à l'autre et même d'un individu à l'autre, recrutant la PrP et la transformant en PrPsc. Les maladies humaines à prion sont très rares. La plus connue est la maladie de Creutzfeld-Jakob dont l'origine peut être sporadique, génétique ou secondaire à une contamination. Son caractère transmissible est à l'origine de mesures de précautions particulières pour l'utilisation de matériels chirurgicaux ou d'exploration endoscopique.
Hypothèse d'une transmission de type « prion » dans les maladies neurodégénératives ?
Depuis quelques années, l'hypothèse d'une propagation prion-like de certaines protéinopathies cérébrales est développée. Selon cette théorie, les protéines mal conformées ont la propriété de pouvoir transmettre leur conformation anormale aux protéines bien conformées, dites « natives », adjacentes. La formation des premiers oligomères correspond à la phase de nucléation (agrégation de la protéine pour former une « graine » ou seed en anglais). La croissance de l'agrégat est assurée par la liaison de monomères natifs et leur conversion conformationnelle pour former des protofibrilles, puis des fibrilles. La fragmentation de la fibrille permet son autopropagation en générant de nouvelles « graines ». Oligomères et fibrilles se transmettraient de cellule en cellule, selon des voies déterminées, ce qui expliquerait la progression spatiotemporelle relativement prédictible des lésions caractérisant certaines maladies (figure 28.3).
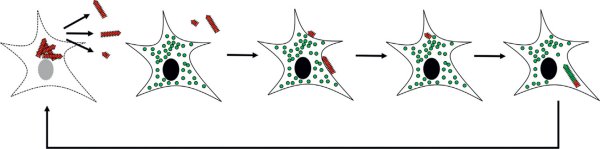
Figure 28.3
Étapes clés dans la propagation de l'alphasynucléine, ou de la protéine tau pathogénique agrégée entre cellules neuronales.
De gauche à droite, l'alphasynucléine, ou la protéine tau, adopte une forme capable de s'agréger (triangles rouges), s'agrège et ces agrégats s'accumulent dans les cellules conduisant à leur mort et leur libération dans le milieu extracellulaire. Ces agrégats se lient à des cellules naïves
contenant de l'alphasynucléine, ou la protéine tau, non pathogénique (cercles verts). Ils sont internalisés par endocytose, recrutent l'alphasynucléine, ou la protéine tau, endogène lorsqu'elle adopte une conformation compatible (triangles verts). Ce faisant, ces agrégats s'amplifient, s'accumulent dans la cellule conduisant à sa mort. Le cycle est ainsi bouclé.
© Melki R. Les protéinopathies infectieuses de Parkinson et d'Alzheimer. Bull Acad Natl Med 2020 ; 204 : 224-31.
Neurodégénérescence et maladies neurodégénératives
Thibaud Lebouvier , Simon Lecerf et Mathieu Ceccaldi
L'un des paradoxes des MND tient dans l'absence de définition consensuelle de leur mécanisme commun, la neurodégénérescence. La physiopathologie complexe des MND, mouvante en fonction de l'avancée des connaissances, a par exemple fait reconnaître l'importance des phénomènes neuro-inflammatoires (cf. chapitre 25) dans le processus neurodégénératif. Ainsi des maladies inflammatoires du système nerveux central (SNC) comme la sclérose en plaques sont-elles dorénavant considérées par certains auteurs comme neurodégénératives. Mais ce débat sur le caractère primitivement ou secondairement neurodégénératif des maladies du SNC et la délimitation précise des MND est secondaire derrière la reconnaissance de mécanismes multiples, intriqués et souvent partagés, suscitant l'espoir d'approches thérapeutiques communes.
Le concept de neurodégénérescence
En l'absence de définition consensuelle, la neurodégénérescence peut être définie comme une dysfonction, puis une perte progressive et irréversible, apparemment spontanée, des neurones et des synapses du système nerveux central. Les frontières entre les MND dans lesquelles la perte neuronale est apparemment « spontanée » et des processus neurodégénératifs secondaires à des affections primitivement inflammatoires, infectieuses ou métaboliques peuvent être ténues.
En fonction des MND, la neurodégénérescence affecte préférentiellement certains neurones et/ou certaines cellules gliales et certaines régions du SNC, déterminant un phénotype clinique qui guide le clinicien pour poser un diagnostic. Ainsi, la vulnérabilité du cortex entorhinal et de l'hippocampe au processus neurodégénératif de la MA explique la prééminence des troubles mnésiques ; la vulnérabilité des neurones dopaminergiques pigmentés de la substance noire au processus neurodégénératif de la maladie de Parkinson explique la survenue du syndrome parkinsonien, etc. Mais au sein de chaque maladie, la topographie de l'atteinte peut varier et rendre le phénotype clinique hétérogène, d'où le recours – quelquefois nécessaire – à des biomarqueurs qui constituent des indicateurs mesurables in vivo d'un processus pathologique (ex : biomarqueurs de MA dans le LCS).
Mécanismes de la neurodégénérescence
Agrégats protéiques et MND
La neurodégénérescence est multifactorielle. Si ses causes et mécanismes précis restent encore incompris, des facteurs génétiques, environnementaux et endogènes liés au vieillissement ont été identifiés. Parmi ces facteurs, les protéinopathies, constituées par l'agrégation de protéines mal conformées, jouent un rôle particulièrement important (cf. supra) et la majorité des MND sont caractérisées sur le plan histologique par les inclusions formées par une ou plusieurs des trois protéines majeures de la neurodégénérescence : la protéine tau, l'α-synucléine, la protéine TDP-43, etc. (cf. supra). Une classification moléculaire des MND reposant sur l'identification de ces marqueurs protéiques supplante actuellement les anciennes classifications cliniques ou anatomiques (cf. tableau 33.4).
Le rôle des agrégats protéiques comme acteurs ou marqueurs passifs du processus dégénératif reste un sujet de débats. La formation des agrégats pourrait plus refléter une réaction protectrice de la cellule ou de l'environnement que le processus dégénératif en lui-même. Néanmoins, la génétique apporte des arguments majeurs en faveur de l'implication des protéines agrégées dans le processus dégénératif car, pour chacune des protéines des agrégats, des mutations sur le gène qui les encode ont été identifiées comme des causes de formes génétiques de MND.
Exemples
- Les mutations sur le gène APP qui encode la protéine précurseur du peptide β-amyloïde sont responsables de formes héréditaires de maladie d'Alzheimer.
- Les mutations sur le gène SCNA qui encode l'α-synucléine sont responsables de formes génétiques de maladies à corps de Lewy.
Les traitements ciblant l'agrégation des protéines sont l'une des voies principales de la recherche sur les MND. Ils visent à diminuer leur production, à empêcher leur agrégation ou à activer la clairance des agrégats (immunothérapies dirigées contre le peptide amyloïde testées dans la MA).
Autres mécanismes
Outre la protéinopathie, d'autres mécanismes sont communs aux processus dégénératifs.
Neuro-inflammation
La neuro-inflammation dans les maladies du SNC est le plus souvent indépendante de l'inflammation périphérique et implique les cellules gliales (microglie et astrocytes) (cf. chapitre 25). Jusqu'à récemment, la conception prévalente était que la neuro-inflammation observée dans les MND (élévation parenchymateuse des cytokines pro-inflammatoires, réaction astro et microgliale) était un phénomène secondaire, réactionnel notamment à la formation des agrégats. Mais l'exemple de la phase secondaire de la sclérose en plaques montre que l'inflammation peut déclencher et nourrir un processus neurodégénératif. Dans la MA, deux facteurs de risque génétiques majeurs, l'allèle ε4 du gène APOE et des variants rares du gène TREM2 codent pour des protéines d'expression essentiellement gliale qui modulent la réponse inflammatoire de la microglie et des astrocytes, plaçant la neuro-inflammation comme acteur majeur du processus dégénératif.
Stress métabolique ou stress oxydant
L'ATP produite par la mitochondrie est indispensable aux fonctions des neurones, de la synthèse des neurotransmetteurs au transport axonal, du maintien des gradients ioniques membranaires à l'organisation des vésicules synaptiques. Dans le neurone, les mitochondries sont présentes dans tous les sites à haute demande d'énergie comme les terminaisons axonales ou les dendrites. L'âge et d'autres facteurs peuvent impacter les fonctions mitochondriales, ce qui provoque une production accrue d'espèces réactives d'oxygène (cf. encadré 28.1), pro-oxydantes. Une balance défavorable entre substances pro-oxydantes et antioxydantes définit le stress oxydant, qui provoque une cascade délétère d'évènements (mutations de l'ADN mitochondrial, neuro-inflammation, dysfonction protéique) (figure 28.4). La dysfonction mitochondriale a été particulièrement étudiée dans la maladie de Parkinson ; certaines mutations responsables de formes génétiques précoces de la maladie affectent des gènes (PINK1 ou PARK2) directement impliqués dans les fonctions mitochondriales.
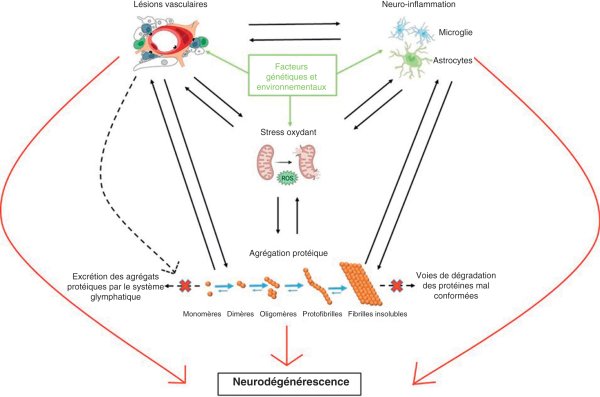
Figure 28.4
Mécanismes de la neurodégénérescence.
Différentes anomalies, telles que les lésions vasculaires, le stress oxydant, la neuro-inflammation, l'agrégation protéique, associées à des facteurs génétiques et environnementaux, sont de manière directe et indirecte à l'origine de la neurodégénérescence.
© Illustration de Simon Lecerf.
Couplage neurovasculaire
Une activité neuronale accrue provoque une augmentation du flux sanguin dans la région concernée : c'est le couplage neurovasculaire, qui est mis à profit en IRM fonctionnelle pour visualiser l'activation d'aires corticales après une tâche cognitive ou sensorimotrice. Ce couplage est assuré par l'unité neurovasculaire, qui est composée par les cellules endothéliales, les péricytes, les cellules musculaires lisses vasculaires et les astrocytes. L'unité neurovasculaire maintient l'intégrité de la barrière hématoencéphalique et contribue au système glymphatique, qui assure la clairance des déchets accumulés dans le liquide interstitiel du parenchyme cérébral (cf. chapitre 25). L'altération de l'unité neurovasculaire semble plus fréquente dans les MND et rend la barrière hématoencéphalique perméable, et l'hypoxie et la neuro-inflammation qui en résultent contribuent à la neurodégénérescence.
Neurodégénérescence et vieillissement normal
Les MND pourraient être théoriquement délimitées par la négative, par ce qu'elles ne sont pas, c'est-à-dire le vieillissement cérébral normal. Néanmoins, cette distinction renvoie à des controverses sur les notions de frontière ou de continuum entre normal et pathologique et à la définition ambiguë de la normalité, qui oscille entre le concept statistique de moyenne et le concept normatif d'idéal. L'analyse histologique des cerveaux de sujets sains montre la prévalence importante des lésions vasculaires et dégénératives avec l'âge. La fréquence de ces lésions apparemment asymptomatiques justifie-t-elle de les considérer comme « normales » ?
L'ensemble des mécanismes ci-dessus ont un effet synergétique entre eux et sur la neurodégénérescence" (cf. figure 28.4).
Vieillissement cérébral normal
En considérant la normalité comme un concept statistique, il est possible d'étudier les modifications cérébrales dans des séries de patients décédés indemnes de MND « clinique ». Il existe d'abord une grande variabilité interindividuelle de l'atrophie cérébrale liée à l'âge. La réduction de volume est tardive et peu marquée, estimée à 2 % par décennie à partir de 50 ans. Le poids moyen de l'encéphale, situé entre 1 300 et 1 400 g avant 60 ans, tend à diminuer au-delà pour atteindre 1 100 g chez les centenaires (données du département de neuropathologie Escourolle, Paris). Les déterminants de l'atrophie sont multiples : l'atrophie du corps cellulaire et de l'arborisation dendritique des neurones et la rétraction du tissu nerveux jouent un rôle ; la perte neuronale intervient vraisemblablement mais est difficile à évaluer. D'autres modifications surviennent en parallèle et peuvent être corrélées au vieillissement cognitif. Des altérations sont décrites sur les grands systèmes neurochimiques (cholinergique, sérotoninergique, dopaminergique, glutamatergique), notamment dans les réseaux préfrontaux. L'âge est associé à une inflammation chronique de bas grade. Enfin l'architecture microvasculaire cérébrale subit des modifications profondes, notamment une perte capillaire qui peut atteindre 15 à 50 % selon les régions, et pourrait être l'un des déterminants de la leucoaraïose (cf. chapitre 27). Ces remaniements de la substance blanche périventriculaire et profonde se manifestent par des hyperintensités T2 qui sont communes en IRM chez le sujet âgé indemne de trouble cognitif ou moteur.
En outre, certaines des lésions rapportées dans les MND sont communes dans les cerveaux de sujets âgés décédés indemnes de MND : des plaques amyloïdes sont retrouvées chez 50 % des sujets « normaux » à l'âge de 74 ans (données du département de neuropathologie Escourolle, Paris).
Frontières entre vieillissement normal et pathologique
Ce sont la densité et l'extension topographique des lésions neurodégénératives qui distinguent le vieillissement cérébral pathologique du vieillissement cérébral normal. La clinique mieux que la pathologie permet de distinguer clairement des trajectoires de déclin cognitif ou moteur qui s'écartent franchement de la trajectoire du vieillissement normal. Ainsi les troubles cognitifs des MND telles que la MA se distinguent du déclin cognitif « normal » du vieillissement qui affecte la vitesse de traitement, les ressources en mémoire de travail et les capacités d'inhibition.
La fréquence des lésions dégénératives chez le sujet âgé et le fait que la plupart des MND sont caractérisées par une longue phase préclinique, où les lésions sont présentes mais ne provoquent pas de symptômes, gênent le neuropathologiste. Les corrélations entre les lésions dégénératives identifiées à l'analyse du cerveau et la clinique sont parfois difficiles à déterminer, et le diagnostic reste alors probabiliste (en termes de probabilité que le phénotype clinique observé du vivant du patient soit lié à la pathologie identifiée, tenant en compte la densité et la répartition des lésions). Des discordances sont parfois observées entre une pathologie neurodégénérative cérébrale intense et étendue et la modestie des manifestations cliniques : le terme de résilience cérébrale désigne ces situations où le fonctionnement cérébral est meilleur qu'attendu compte tenu des lésions observées, qui seraient en partie liées à une meilleure réserve cognitive. Ce terme de réserve cognitive désigne l'ensemble des caractéristiques qui protègent contre le déclin cognitif lié à l'âge, telles le niveau d'éducation, l'activité professionnelle, les activités de loisirs et les interactions sociales tout au long de la vie. Les stratégies pour améliorer la réserve cognitive et la résilience cérébrale sont une autre voie stimulante de recherche contre les MND.
- La neurodégénérescence est une dysfonction puis une perte progressive et irréversible, apparemment spontanée, des neurones et des synapses du système nerveux central.
- Les agrégats protéiques jouent un rôle majeur dans le processus dégénératif, assimilant la plupart des maladies neurodégénératives à des protéinopathies.
- Une classification moléculaire des protéinopathies est possible en fonction de la nature des agrégats protéiques. La majorité des MND sont caractérisées par les agrégats formés par une ou plusieurs parmi quatre protéines majeures : protéine tau, α-synucléine, protéine TDP-43 et peptide β-amyloïde.
- Des mutations sur le gène encodant ces protéines peuvent causer des formes génétiques de maladies neurodégénératives.
- L'agrégation survient à la suite de modifications conformationnelles conduisant à des agrégats de protéines visibles en histologie ; ces derniers saturent les systèmes cellulaires de clairance des protéines anormales (système ubiquitine-protéasome et lysosomes responsables de l'autophagie).
- L'hypothèse d'une transmission de type prion est mise en avant pour expliquer la propagation de la protéinopathie.
- La pathologie vasculaire cérébrale pourrait promouvoir le processus dégénératif en altérant l'unité neurovasculaire.
- Vieillissement cérébral normal et maladies neurodégénératives se situent sur un continuum. C'est la densité et l'extension topographique des lésions neurodégénératives et leur corrélation à des manifestations cliniques évolutives qui caractérisent les maladies neurodégénératives.
Voir QRM chapitre 32.
Pour en savoir plus
- Bezard E, Dehay B. Maladie de Parkinson. Le rôle de la synucléine. Med/Sci 2022 ;38:45–51.
- Breijyeh Z, Karaman R. Comprehensive review on Alzheimer's disease : causes and treatment. Molecules 2020 ;25:5789.
- Haik S. Les mécanismes de type prion dans les maladies neurodégénératives. La lettre du neurologue 2017 ;10:318–22.
- Kempermann G, Gage FH, Aigner L, et al. Human adult neurogenesis : Evidence and remaining questions. Cell Stem Cell 2018 ;23:25–30.