Rappelons que les voies motrices sont des voies descendantes qui transmettent la commande motrice cérébrale au muscle. Le mouvement est sous un contrôle volontaire dépendant de la voie corticospinale. L'exécution du mouvement implique une régulation semi-volontaire et automatique dépendant des voies des ganglions de la base. La coordination des mouvements et de l'équilibre est sous le contrôle cérébelleux, soit directement, soit indirectement via ses nombreuses afférences. D'autres systèmes contribuent au bon déroulement du mouvement, notamment le système vestibulaire et la sensibilité profonde consciente.
Motricité volontaire
Bastien Joubert , Bruno Brochet Relecteurs : et Elisabeth Ruppert
Rappel anatomophysiologique (cf. chapitre 1)
La motricité volontaire est commandée par les aires motrices primaires et secondaires situées au niveau du lobe frontal du cerveau. L'exécution du mouvement est sous la commande de l'aire motrice primaire, qui occupe le gyrus précentral, tandis que sa programmation et sa planification font intervenir les aires motrices secondaires (cortex prémoteur et aire motrice supplémentaire) situées juste en avant de l'aire motrice primaire.
L'aire motrice primaire prend en charge la motricité de l'hémicorps controlatéral et suit une organisation somatotopique : la face est représentée dans sa partie inférolatérale, le membre supérieur dans sa partie supérolatérale, et le membre inférieur dans sa partie médiane. Les neurones de l'aire motrice primaire projettent des axones qui constituent deux faisceaux : le faisceau corticospinal (anciennement dénommé faisceau pyramidal) à destination de la corne ventrale de la moelle spinale, et le faisceau corticonucléaire (appelé précédemment faisceau géniculé) vers les noyaux moteurs des nerfs crâniens. À leur origine, les fibres des faisceaux corticospinal et corticonucléaire sont dispersées dans la substance blanche des hémisphères cérébraux, puis elles descendent et se regroupent au niveau de la capsule interne (bras postérieur pour le faisceau corticospinal, genou pour le faisceau corticonucléaire). Une lésion touchant la capsule interne, où les fibres corticospinales sont densément regroupées, provoque donc un déficit moteur proportionnel controlatéral, aux trois étages (face, membre supérieur et membre inférieur), tandis qu'une lésion de même volume touchant le cortex cérébral ou le centre ovale (corona radiata) des hémisphères, où les fibres sont dispersées, provoque un déficit moteur prédominant sur une partie de l'hémicorps controlatéral. Ayant traversé la capsule interne, le faisceau corticospinal descend dans les pédoncules cérébraux à la partie ventrale du mésencéphale, traverse le pont où il est dissocié par les noyaux de celui-ci, puis se regroupe à nouveau au sein des pyramides bulbaires. Au niveau bulbaire, la majorité des fibres décussent pour constituer du côté opposé le faisceau corticospinal croisé de la moelle spinale. Une minorité de fibres reste du côté homolatéral pour former le faisceau corticospinal direct, dont la décussation est étagée sur les niveaux médullaires successifs.
Les deuxièmes neurones moteurs (anciennement dénommés motoneurones α) situés dans la corne ventrale et dans les noyaux des nerfs crâniens reçoivent les projections des voies corticospinale et corticonucléaire, respectivement, et envoient à leur tour des axones qui passent par les nerfs périphériques ou crâniens pour aller se projeter sur les fibres musculaires. Chaque 2e neurone moteur unique innerve plusieurs fibres musculaires au sein d'un muscle, constituant avec elles une unité motrice. La transmission neuromusculaire s'effectue par une synapse entre une terminaison axonale du 2e neurone moteur et une plaque motrice, qui est une zone spécialisée à la surface des fibres musculaires contenant des récepteurs à l'acétylcholine. La libération d'acétylcholine par la terminaison axonale provoque la dépolarisation membranaire de la fibre musculaire, ce qui déclenche sa contraction.
La contraction de la fibre musculaire est régulée par un arc réflexe mettant en jeu les fuseaux neuromusculaires, qui sont des structures sensibles à l'étirement et présentes entre les fibres musculaires. L'étirement d'un fuseau neuromusculaire active un neurone sensitif de la moelle spinale qui se projette à son tour sur un 2e neurone moteur, provoquant de manière réflexe la contraction de l'unité motrice. Cet arc réflexe est à l'origine des réflexes ostéotendineux (ROT), qui sont une contraction musculaire brève et immédiate déclenchée par la percussion du tendon du muscle. D'autres réflexes impliquent des arcs réflexes plus complexes, polysynaptiques, comme les réflexes cutanéomuqueux, qui sont des réflexes d'évitement face à un stimulus cutané potentiellement délétère, ou des actions automatiques facilitant la locomotion et l'équilibre. Certains de ces réflexes ont une utilité clinique, comme le réflexe cutané plantaire et les réflexes cutanés abdominal.
La fatigabilité est un déficit moteur fluctuant, apparaissant à la répétition des contractions musculaires. Elle est caractéristique du syndrome myasthénique, qui signe un trouble de la transmission neuromusculaire, soit par défaut de libération d'acétylcholine (syndrome myasthénique présynaptique) soit par manque de récepteurs à acétylcholine (syndrome myasthénique post-synaptique).
Examen clinique
L'examen de la motricité comporte l'évaluation de la force musculaire, du tonus musculaire, de la trophicité et des réflexes. L'examen doit être comparatif entre la droite et la gauche.
La force musculaire s'apprécie d'abord de façon globale, en demandant au patient de maintenir une posture contre la gravité pendant au moins 10 secondes ; le déficit moteur est révélé par la chute du membre. On utilise la manœuvre de Barré aux membres supérieurs (bras tendus en avant), la manœuvre de Barré aux membres inférieurs (patient en décubitus ventral, genoux fléchis à 90°, jambes maintenues à la verticale), et la manœuvre de Mingazzini (décubitus dorsal, cuisses maintenues à la verticale et jambes à l'horizontale, genoux fléchis). La motricité est ensuite évaluée de façon segmentaire en demandant au patient d'exercer un effort contre résistance, groupe musculaire par groupe musculaire. Le déficit est coté sur une échelle de 0 à 5 (tableau 8.1).
Tableau 8.1.
Cotation de la force musculaire.
| 0 |
Aucune contraction musculaire |
| 1 | Contraction musculaire sans mouvement dans le plan du lit |
| 2 | Mouvement dans le plan du lit, pas contre la gravité |
| 3 | Mouvement contre gravité, pas contre résistance |
| 4 | Mouvement contre résistance |
| 5 | Force musculaire normale |
Publié avec l'aimable autorisation de Medical Research Council © Crown copyright.On recherche une hypertonie musculaire en mobilisant passivement les segments des membres ; faire varier la vitesse de la mobilisation permet de distinguer l'hypertonie élastique (spasticité) d'origine pyramidale, de l'hypertonie plastique (rigidité) d'origine extrapyramidale (cf. infra). En distalité, le tonus est évalué en imprimant un mouvement de rotation (poignet, cheville). On recherche une hypotonie en imprimant un mouvement passif ample à la partie proximale des membres : l'amplitude du ballant du membre est augmentée du côté hypotone.
Trois réflexes cutanéomuqueux et six ROT sont recherchés (tableau 8.2). Les ROT sont recherchés par la percussion du tendon du muscle au repos et la réponse est jugée sur la contraction du muscle. On peut sensibiliser le test en demandant au sujet de contracter des groupes musculaires distants (ex : serrer les poings le plus fort possible).
Tableau 8.2.
Examen des réflexes.
| Réflexe | Méthode | Résultat | Niveau radiculaire | Niveau tronculaire |
|---|---|---|---|---|
| Réflexes cutanéomuqueux | ||||
| Réflexe cutané plantaire | Stimulation du bord latéral de la plante du pied d'arrière en avant | Flexion de l'hallux et creusement de la voûte plantaire | S1-S2 | |
| Réflexe cutané abdominal | Effleurement transversal de la paroi abdominale à droite et à gauche de la ligne médiane, au-dessus, au niveau et au-dessous de l'ombilic (réflexe cutané abdominal supérieur, moyen et inférieur) | Brève contraction de la paroi abdominale avec attraction de l'ombilic | Supérieur T6-T8 Moyen T8-T10 Inférieur T10-T12 |
|
| Réflexe crémastérien | Stimulation de la face interne de la cuisse | Ascension du testicule Rétraction de la grande lèvre |
L1-L2 | |
| Réflexes ostéotendineux | ||||
| Réflexe bicipital | Avant-bras en semi-flexion et supination Percussion du pouce de l'examinateur placé sur le tendon du biceps brachial du patient |
Contraction du biceps et flexion du coude | C5 | Nerf musculocutané |
| Réflexe styloradial | Avant-bras en semi-flexion et en position intermédiaire entre la pronation et la supination Percussion du bord latéral du radius un peu au-dessus du processus styloïde radial |
Contraction du brachioradial et flexion du coude | C6 | Nerf radial |
| Réflexe tricipital | Avant-bras en semi-flexion maintenu par l'examinateur Percussion du tendon du triceps brachial au-dessus de l'olécrâne |
Contraction du triceps et extension du coude | C7 | Nerf radial |
| Réflexe ulnopronateur | Avant-bras en semi-flexion et en légère supination Percussion du processus styloïde ulnaire |
Pronation de l'avant-bras | C8 | Nerf ulnaire |
| Réflexe patellaire | Percussion du tendon patellaire chez un patient assis jambes pendantes, ou bien couché jambe semi-fléchie soutenue par l'examinateur | Contraction du quadriceps et extension du genou | L4 | Nerf fémoral |
| Réflexe calcanéen | Percussion du tendon calcanéen, pied maintenu en flexion dorsale par l'examinateur | Contraction du triceps sural et flexion plantaire du pied | S1 | Nerf tibial |
Sémiologie
La distribution du déficit moteur et les anomalies du tonus musculaire, des réflexes, et de la trophicité musculaire permettent d'évaluer si la lésion en cause porte atteinte :
- à la voie corticospinale, sur son trajet entre les corps cellulaires des premiers neurones moteurs dans le cortex cérébral et leur axone constituant le faisceau corticospinal, responsable d'un syndrome pyramidal ;
- au 2e neurone moteur situé dans la corne ventrale de la moelle spinale et dont l'axone emprunte la racine ventrale du nerf spinal, puis le nerf spinal et ses branches ventrale et dorsale jusqu'aux nerfs périphériques, responsable d'un syndrome neurogène ;
- à la jonction neuromusculaire, responsable d'un syndrome myasthénique ;
- au muscle, responsable d'un syndrome myogène (tableau 8.3). En particulier, les ROT sont abolis en cas d'atteinte des neurones moteurs effecteurs (neuropathie motrice) ou de déafférentation sensitive périphérique (neuropathie sensitive). Ils sont au contraire anormalement vifs en cas de déafférentation des neurones moteurs de la voie corticospinale.
Tableau 8.3.
Caractéristiques des différents types de déficit moteur.
| Distribution du déficit moteur | Réflexes ostéotendineux | Réflexes cutanéomuqueux | Tonus musculaire | Trophicité musculaire | |
|---|---|---|---|---|---|
| Lésion de la voie corticospinale | Hémicorporel si lésion unilatérale | Vifs, exagérés, polycinétiques (parfois abolis à la phase aiguë) | Abolis | Hypertonie élastique (parfois hypotonie à la phase aiguë) | Normale |
| Atteinte neurogène périphérique | Variable | Absents ou diminués | Abolis | Normal ou hypotonie | Hypotrophie |
| Atteinte de la jonction neuromusculaire | Variable, forme oculomotrice | Normaux | Normaux | Normal | Normale |
| Atteinte myogène | Bilatéral et proximal | Normaux | Normaux | Normal | Hypo ou hypertrophie |
Syndrome pyramidal : sémiologie d'une atteinte des voies corticospinales
Le syndrome pyramidal regroupe l'ensemble des symptômes et signes cliniques résultant de l'atteinte, à quelque niveau que ce soit, de la voie corticospinale (anciennement dénommée voie pyramidale), support de la commande motrice volontaire.
Le syndrome pyramidal est caractérisé par l'association de signes déficitaires qui traduisent l'atteinte du faisceau corticospinal, et de signes de spasticité, liés à la libération d'activités motrices réflexes normalement inhibées par la voie corticospinale. La survenue de ces deux composantes peut être synchrone en cas d'atteinte lentement progressive ou décalée dans le temps en cas d'atteinte aiguë. Dans ce dernier cas, la paralysie est d'abord flasque (hypotonie), puis spastique (hypertonie pyramidale).
Le syndrome pyramidal est controlatéral à la lésion lorsque celle-ci siège au-dessus du bulbe, homolatéral à la lésion lorsque celle-ci est médullaire.
Symptômes
- La faiblesse motrice est d'intensité et de topographie variables. Les malades peuvent utiliser des termes comme paralysie, gêne, ankylose, lourdeur, maladresse, mauvais équilibre, etc. Elle retentit sur les activités motrices courantes telles que marcher, courir, gravir les escaliers, se relever d'un siège, porter des charges lourdes, etc.
- La claudication motrice intermittente est évocatrice d'un syndrome pyramidal. Le patient se plaint d'une faiblesse indolore d'un ou des deux membres inférieurs, survenant après un temps de marche, disparaissant au repos pour réapparaître par la suite. Ce symptôme s'observe surtout en cas d'atteinte médullaire.
- Des sensations de contractures et/ou de raideur peuvent s'exprimer comme douloureuses, liées à la spasticité.
- Des troubles de la phonation et de la déglutition peuvent s'observer en cas d'atteinte bilatérale (syndrome pseudobulbaire).
- Des mictions impérieuses, le malade ne pouvant se retenir d'uriner, traduisent une vessie spastique.
Signes cliniques
Déficit moteur lié à un déficit de la commande motrice volontaire
- En cas de lésion aiguë et étendue, le déficit est massif, concernant toute la musculature, de topographie variable selon la localisation de la lésion. Il touche par exemple un hémicorps comprenant face, membre supérieur et membre inférieur, ou deux membres inférieurs, plus rarement un seul membre.
- En cas de lésion partielle ou progressive, le déficit prédomine sur certains groupes musculaires.
- Au membre supérieur, le déficit musculaire prédomine sur les muscles extenseurs et entraîne l'atteinte des mouvements fins et rapides des doigts. On décrit trois signes cliniques :
- le signe de Barré aux membres supérieurs à l'épreuve des bras tendus (les bras sont tendus à l'horizontale) : on observe une chute progressive du membre supérieur concerné, le déficit atteint tout le bras, parfois seulement le poignet ou les doigts ;
- le signe de la main creuse de Garcin, à l'épreuve des bras fléchis et des doigts fortement écartés, on observe que la main du patient se creuse progressivement par déficit des extenseurs (« main creuse ») ;
- une lenteur des mouvements alternatifs rapides comme l'opposition pouce-index, en comparant un côté à l'autre.
- Au membre inférieur, le déficit musculaire prédomine sur les muscles fléchisseurs. On décrit deux signes cliniques :
- le signe de Mingazzini : lorsque le patient est installé en décubitus dorsal et garde les cuisses fléchies et les jambes maintenues à l'horizontale,
- le déficit s'extériorise par la chute progressive du membre inférieur (cuisse et/ou jambe) ;
- le signe de Barré aux membres inférieurs : lorsque le patient est installé en décubitus ventral et garde les jambes fléchies à 90°, le déficit s'extériorise par la chute progressive de la jambe.
- Au niveau de la face, sur le territoire facial inférieur, on observe :
- un effacement du pli nasolabial ;
- une asymétrie lorsqu'on demande au patient de découvrir les dents, le territoire facial supérieur étant respecté car il reçoit des fibres de chaque hémisphère cérébral. Dans les formes discrètes, l'asymétrie au niveau du territoire facial inférieur se démasque aux mouvements automatiques comme le fait de compter rapidement, alors qu'elle n'est pas visible au sourire forcé volontaire par exemple, ce qui est connu sous le nom de dissociation automaticovolontaire ;
- une dysarthrie paralytique avec voix nasonnée et troubles de la déglutition dans le cadre d'un syndrome pseudobulbaire par atteinte des voies corticonucléaires.
- Au membre supérieur, le déficit musculaire prédomine sur les muscles extenseurs et entraîne l'atteinte des mouvements fins et rapides des doigts. On décrit trois signes cliniques :
Troubles du tonus musculaire
- En cas de lésion aiguë et étendue, on observe une hypotonie. Le déficit musculaire est flasque.
- En cas de lésion partielle ou progressive, ou bien quelques semaines après la phase hypotonique, on observe une hypertonie pyramidale, appelée spasticité, caractéristique du syndrome pyramidal. La spasticité est une contracture musculaire ayant plusieurs caractères :
- la topographie au membre supérieur où elle prédomine sur les fléchisseurs pouvant imprimer une atteinte permanente en flexion du coude, du poignet et des doigts ;
- la topographie au membre inférieur où elle prédomine sur les extenseurs avec jambe en extension, pied en varus équin, démarche en fauchant avec une marche spastique, pied « raclant » le sol avec sa pointe et son bord externe ;
- son accentuation à l'action, pouvant n'apparaître, par exemple, qu'après un certain temps de marche en cas de claudication motrice intermittente ;
- son accentuation avec l'angle et la vitesse d'étirement ;
- son élasticité, cédant comme une lame de canif, contrairement à l'hypertonie extrapyramidale, qui est dite plastique ;
- sa douleur éventuelle quand elle est intense.
Anomalies des réflexes ostéotendineux et cutanés
Les anomalies des ROT varient en fonction de l'étendue et de la vitesse d'installation du déficit moteur. En cas de lésion aiguë et étendue, on observe une aréflexie ostéotendineuse. En cas de lésion progressive ou partielle ou bien à distance d'une lésion aiguë, on observe une hyperréflexie ostéotendineuse. Les ROT sont alors vifs, diffusés, polycinétiques. La vivacité désigne l'ampleur excessive de la réponse motrice. La diffusion peut être celle de la réponse motrice, lorsque celle-ci s'observe aussi sur d'autres segments du même membre ou sur le membre controlatéral ou celle de la zone réflexogène lors de la percussion avec le marteau à réflexes, par exemple l'obtention de la réponse après percussion de la crête tibiale pour le réflexe patellaire, ou l'obtention du réflexe médioclaviculaire, qui est pathologique. On l'obtient à la percussion avec le marteau à réflexes du doigt de l'examinateur posé sous la clavicule. En cas normal, cette manœuvre n'entraîne pas de réponse, en cas de syndrome pyramidal, on obtient une réponse motrice de l'ensemble du membre supérieur.
Le polycinétisme signifie que plusieurs réponses motrices se succèdent jusqu'à entraîner un clonus inépuisable de la patella ou du pied (« trépidation épileptoïde » du pied). Le clonus du pied/de la cheville est le meilleur signe de spasticité. Il se recherche en exerçant une dorsiflexion brusque du pied et en maintenant cette position. Le pied est alors animé de mouvements brefs et rythmés de flexion-extension, typiquement inépuisables.
Le signe de Babinski correspond à une réponse en extension de l'hallux et écartement des autres orteils lors de la recherche du réflexe cutané plantaire. Normalement, une stimulation d'arrière en avant, du bord externe de la plante du pied, résulte en une flexion de l'hallux. Parfois, un stimulus cutané quelconque peut déclencher un signe de Babinski.
D'autres signes cliniques ont une moindre valeur :
- les syncinésies, qui sont des mouvements involontaires ou des renforcements toniques survenant dans des groupes musculaires, lors de mouvements volontaires concernant une autre partie du corps. Par exemple, chez un malade faisant les marionnettes avec la main droite, la main gauche ébauche le même mouvement ;
- le signe d'Hoffmann avec apparition d'une flexion des doigts lors d'une pression vive sur l'ongle du majeur ;
- une abolition des réflexes cutanés abdominaux.
Chez certains sujets, dits « neurotoniques », les ROT peuvent être vifs, parfois même polycinétiques avec une ébauche de clonus du pied, épuisable, sans que cela témoigne nécessairement d'un syndrome pyramidal. L'extension de la zone réflexogène n'est jamais observée dans ce cas.
Syndrome pyramidal
- Le déficit moteur prédomine dans le syndrome pyramidal aux extenseurs du membre supérieur, aux fléchisseurs du membre inférieur et au territoire inférieur du nerf facial.
- La spasticité ou hypertonie pyramidale prédomine sur les fléchisseurs du membre supérieur, sur les extenseurs du membre inférieur avec une démarche « en fauchant », est élastique, accrue à l'action.
- Les ROT sont vifs, diffusés (extension de la zone réflexogène), polycinétiques (clonus du pied).
- Un signe de Babinski avec extension de l'hallux est présent lors de la recherche du réflexe cutané plantaire.
Syndrome neurogène périphérique : sémiologie d'un déficit musculaire consécutif à une atteinte neurogène périphérique
Le syndrome neurogène périphérique regroupe l'ensemble des symptômes et signes cliniques qui témoignent d'une atteinte du SNP comprenant :
- un neurone moteur dont le corps cellulaire est situé dans la corne ventrale de la moelle spinale ;
- un neurone sensitif dont le corps cellulaire est situé dans le ganglion spinal ;
- un contingent végétatif, ou autonome.
Symptômes
- La faiblesse traduisant le déficit moteur et pouvant concerner un ou plusieurs membres est décrite par des termes comme maladresse, gêne, lourdeur, etc.
- Les troubles sensitifs peuvent être divers. Les symptômes sensitifs décrits sont :
- des douleurs de type décharges électriques, brûlures, crampes au repos ;
- des paresthésies de type fourmillements, picotements ;
- une anesthésie ou une hypoesthésie, « comme après une anesthésie locale chez le dentiste », une sensation de « peau cartonnée », etc.
Signes cliniques
- Le déficit moteur est de topographie variable, mais prédomine le plus souvent en distal. Il est responsable d'un trouble de la marche dénommée « steppage ». Le déficit moteur est hypotonique et son intensité variable, qu'il est possible de coter, muscle par muscle (cf. tableau 8.1).
- L'amyotrophie est un signe majeur du syndrome neurogène périphérique, mais elle est habituellement absente au début de l'atteinte.
- Les fasciculations correspondent à des contractions musculaires très brèves, superficielles, localisées à une partie d'un muscle, ne déplaçant pas le segment de membre, bien visibles à jour frisant, survenant spontanément ou après percussion du muscle. Elles sont d'une grande valeur sémiologique, quasi pathognomonique, mais très inconstante. Elles sont surtout présentes lors de l'atteinte des neurones moteurs des cornes ventrales de la moelle spinale.
- Les ROT sont abolis ou diminués. Il s'agit, avec l'amyotrophie, du meilleur signe clinique du syndrome. Cependant, c'est un signe clinique inconstant, notamment quand le tronc nerveux ou la racine atteints n'est pas impliqué dans un arc réflexe cliniquement accessible.
- Le déficit sensitif peut toucher la sensibilité protopathique (froid, chaud, piqûre) par atteinte des petites fibres myéliniques et des fibres amyéliniques, la sensibilité épicritique avec déficit au tact superficiel et la sensibilité profonde (sens de position de l'hallux, perception des vibrations du diapason) par atteinte des grosses fibres myéliniques. Le déficit sensitif manque lors des atteintes électives de la corne ventrale de la moelle spinale. L'atteinte sensitive est davantage développée dans le chapitre 7.
- Les troubles trophiques et signes végétatifs sont caractérisés par :
- une peau sèche, amincie, dépilée avec troubles vasomoteurs ;
- des maux perforants plantaires, rétractions tendineuses, pieds creux, rarement arthropathies ;
- des signes de dysautonomie dans certaines neuropathies touchant le système nerveux végétatif avec alors une hypotension orthostatique, une dysfonction érectile et des troubles vésicaux.
Syndrome neurogène périphérique
- Le déficit moteur est de topographie variable, mais le plus souvent distal avec une amyotrophie généralement différée dans le temps, des fasciculations inconstantes et une aréflexie ostéotendineuse.
- Les troubles sensitifs décrits sont « subjectifs », de type douleurs, paresthésies et/ou « objectifs » avec une hypoesthésie ou une anesthésie.
- Des troubles végétatifs sont également possibles.
Syndrome myasthénique : sémiologie d'une atteinte de la jonction neuromusculaire
Le syndrome myasthénique regroupe l'ensemble des symptômes et des signes cliniques résultant d'un dysfonctionnent de la jonction (ou synapse) neuromusculaire.
Symptômes
Le maître symptôme est la fatigabilité musculaire. Il s'agit d'un déficit moteur lié à l'effort et s'améliorant ou disparaissant au repos, d'où la fluctuation des symptômes dans la journée, leur recrudescence en fin de journée et, plus encore, leur caractère intermittent. Les symptômes peuvent être totalement absents au repos, notamment le matin au réveil. Le déficit moteur peut atteindre tous les muscles striés de l'organisme. Plusieurs domaines musculaires sont particulièrement sensibles :
- l'atteinte oculomotrice est caractérisée par :
- un ptosis, qui est une chute de la paupière supérieure, asymétrique, éventuellement alternant (droit puis gauche) avec, typiquement, le signe compensateur du sourcil en lien avec une hypercontraction du muscle frontal,
- une diplopie : le patient décrit une vision double,
- une motilité pupillaire toujours respectée ;
- l'atteinte de la phonation, de la déglutition et de la mastication est caractérisée par :
- une voix nasonnée, surtout en fin de conversation,
- une déglutition difficile, surtout en fin de repas, avec parfois des fausses routes alimentaires et un risque de pneumopathie d'inhalation ou de reflux alimentaire par le nez,
- une mastication déficitaire, surtout en fin de repas ;
- l'atteinte des membres est caractérisée par une faiblesse proximale ;
- l'atteinte des muscles de la nuque est caractérisée par une tête qui fléchit après un temps de marche associée parfois à des cervicalgies ;
- l'atteinte respiratoire par paralysie des muscles intercostaux et du diaphragme est caractérisée par une polypnée superficielle pouvant aller jusqu'à l'asphyxie. Le pronostic vital peut donc être compromis, il s'agit d'une urgence médicale nécessitant une surveillance et prise en soins en milieu de réanimation.
Il n'y a jamais de symptomatologie sensitive de type douleurs ou paresthésies car la synapse neuromusculaire, uniquement motrice, est la seule structure atteinte.
Signes cliniques
- L'examen neurologique peut être strictement normal s'il est pratiqué à distance de tout effort. Le syndrome myasthénique peut être localisé à un groupe de muscles, comme dans les formes oculaires pures, ou être généralisé.
- Un déficit moteur peut apparaître à l'examen clinique après un effort répété, intense, par exemple l'apparition d'un ptosis après une dizaine d'accroupissements.
- Un déficit moteur permanent peut exister, notamment en regard :
- des racines des membres inférieurs, le sujet étendu sur le dos ne peut tenir les membres inférieurs fléchis plus d'une minute ;
- des muscles fléchisseurs de la nuque ;
- des muscles orbiculaires des paupières.
- Dans tous les cas, plusieurs signes négatifs sont à noter :
- absence d'amyotrophie et de fasciculations ;
- absence de modification des ROT ;
- absence de déficit sensitif.
Syndrome myasthénique
- Le syndrome myasthénique est caractérisé par une fatigabilité fluctuante, c'est un déficit moteur lié à l'effort et disparaissant au repos. L'examen clinique réalisé au repos peut être normal.
- L'atteinte est fréquemment oculomotrice avec ptosis et diplopie.
- La gravité potentielle de l'atteinte respiratoire justifie d'une surveillance et d'une prise en soins en réanimation.
Syndrome myogène : sémiologie d'une atteinte musculaire
Le syndrome myogène regroupe l'ensemble des symptômes et signes cliniques résultant d'une myopathie, c'est-à-dire d'une maladie du muscle lui-même.
Symptômes
- La faiblesse musculaire retentit sur les activités motrices courantes telles que marcher, courir, gravir les escaliers, se relever d'un siège, porter des charges lourdes, etc.
- Des douleurs musculaires, myalgies, et des crampes avec contraction en boule d'un muscle, accompagnent la faiblesse musculaire. Elles sont déclenchées ou non par les efforts.
Signes cliniques
- Le déficit moteur est proximal et surtout bilatéral. Il prédomine à la racine des membres et sur la musculature axiale.
- Une atteinte de la ceinture pelvienne et des muscles paravertébraux résulte en :
- une marche dandinante, « en canard » ;
- des difficultés à se relever de la position accroupie ;
- des difficultés à se relever de la position assise ou signe du tabouret. Lors du « relever myopathique », le malade prend appui avec ses mains sur les genoux et « grimpe » le long de ses cuisses ;
- une hyperlordose par atteinte des muscles paravertébraux.
- Une atteinte de la ceinture scapulaire et de la nuque résulte en :
- un déficit des muscles deltoïdes, des biceps et triceps brachiaux ;
- une scapula alata, qui est un décollement des scapulas par paralysie des grands dentelés ;
- un déficit des fléchisseurs de la nuque.
- D'autres muscles peuvent être atteints, plus rarement :
- les muscles de la face avec déficit des muscles orbiculaires des paupières, ptosis suite à un déficit du releveur de la paupière supérieure et des muscles oculomoteurs ou pharyngolaryngés ;
- les muscles distaux ;
- les muscles respiratoires avec un syndrome respiratoire restrictif ;
- le cœur avec une cardiomyopathie.
- Une atteinte de la ceinture pelvienne et des muscles paravertébraux résulte en :
- Le déficit moteur est d'intensité variable selon le degré d'évolution de la myopathie (cf. tableau 8.3).
- Les modifications du volume musculaire sont généralement une amyotrophie, plus rarement une hypertrophie. L'amyotrophie est de même topographie que le déficit moteur, proximal et bilatéral, et de sévérité variable. Elle peut parfois être masquée par le pannicule adipeux ou être absente dans les myopathies métaboliques. L'hypertrophie, plus rare, concerne surtout les mollets.
- Des anomalies de la contraction ou de la décontraction musculaire sont observées dans le syndrome myogène. On note une abolition du réflexe idiomusculaire, c'est-à-dire une absence de contraction. La recherche du réflexe idiomusculaire consiste en la percussion directe du muscle avec un marteau à réflexes. On ne provoque pas la réponse normale qu'est la contraction en masse du muscle suivie d'une décontraction rapide, mais une contraction anormale, « en boules ». La myotonie est caractéristique du syndrome myogène avec une lenteur de la décontraction musculaire, indolore. Elle peut être :
- Spontanée : le patient desserre lentement un objet ou la main de l'examinateur ;
- provoquée par percussion de l'éminence thénar, ce qui provoque la mise du pouce en adduction et un retour lent à sa position initiale.
La myotonie est inconstante, elle ne s'observe que dans certaines myopathies.
- Des rétractions tendineuses peuvent également être présentes avec l'évolution de la maladie.
- Parmi les signes cliniques négatifs, on retient surtout :
- l'absence de déficit sensitif ;
- l'absence d'abolition des ROT, sauf à un stade évolué, quand l'amyotrophie ne permet plus d'obtenir la réponse ;
- l'absence de fasciculations.
Attention
Syndrome myogène 1
Le déficit moteur et l'amyotrophie sont communs au syndrome myogène et au syndrome neurogène périphérique. En faveur du syndrome myogène, on retient :
- un déficit proximal et bilatéral ;
- une abolition du réflexe idéomusculaire ;
- des ROT conservés ;
- une absence de déficit sensitif ;
- une absence de fasciculations.
Le diagnostic syndromique peut être difficile devant certaines amyotrophies d'inutilisation liées à un alitement prolongé, des atteintes du système nerveux central ou une dénutrition.
Syndrome myogène 2
- Dans le syndrome myogène, le déficit moteur est proximal et axial, parfois facial, associé à une amyotrophie de même topographie, plus rarement une hypertrophie du muscle.
- La myotonie correspond à une lenteur à la décontraction musculaire, elle est inconstante.
- Les signes cliniques négatifs permettent de le distinguer du syndrome neurogène périphérique par l'absence de déficit sensitif et la présence des ROT.
Motricité semi-volontaire et automatique
Sara Meoni , Olivier Detante , Bruno Brochet Relecteurs : et Elisabeth Ruppert
Rappel anatomophysiologique (cf. chapitre 1)
La motricité volontaire, sous la commande du cortex moteur primaire, est régulée de façon sous-corticale par les ganglions de la base (autrement appelés noyaux gris centraux) via les voies directes et indirectes. Les ganglions de la base sont des amas de neurones distribués dans la substance blanche du cerveau. Ils participent à l'apprentissage des activités motrices stéréotypées (écriture, vélo, etc.) et facilitent le mouvement en focalisant des informations issues de vastes régions corticales vers l'aire motrice supplémentaire. Ils jouent aussi un rôle de filtre bloquant la réalisation de mouvements inadaptés : un dysfonctionnement des ganglions de la base peut ainsi être à l'origine de mouvements anormaux parasites involontaires. Les ganglions de la base comprennent le noyau caudé, le noyau lentiforme (constitué du globus pallidus interne, du globus pallidus externe et du putamen), le noyau subthalamique, le nucleus accumbens, et la substance noire (qui comprend une partie compacte et une partie réticulaire). Le noyau caudé et le putamen forment un ensemble fonctionnel appelé striatum, qui reçoit de nombreuses afférences corticales et forme la porte d'entrée du système. La sortie est constituée du pallidum, qui envoie des efférences sur l'aire motrice supplémentaire via le thalamus. L'information traverse donc le circuit en passant par les structures suivantes : cortex → striatum → pallidum → thalamus → aire motrice supplémentaire. En parallèle, les neurones à dopamine de la partie compacte de la substance noire ont une influence majeure sur ce réseau en se projetant sur le striatum. On parle de voie nigrostriée dopaminergique.
Examen clinique
Le dysfonctionnement des ganglions de la base, selon le niveau de l'atteinte, peut être à l'origine de différents troubles du mouvement, soit un ralentissement du mouvement (akinésie) avec hypertonie plastique (rigidité), caractéristique du syndrome parkinsonien, soit de mouvements anormaux involontaires. L'analyse des mouvements anormaux doit prendre en considération leur topographie (caractère bilatéral ou non, symétrie, caractère proximal ou distal), leur amplitude, leur caractère rythmique ou non et, le cas échéant, leur fréquence.
L'hypertonie doit être recherchée par la mobilisation passive des membres, et l'akinésie est recherchée en demandant au patient d'effectuer une séquence de mouvements (ouvrir et fermer le poing, battre la mesure avec le pied) le plus rapidement possible, en comparant la rapidité et l'amplitude d'exécution de chaque côté.
Sémiologie
Le syndrome extrapyramidal est, en pratique, un ensemble de syndromes moteurs où l'on classe d'une part le syndrome parkinsonien, d'autre part les mouvements anomaux involontaires.
Le syndrome parkinsonien est dû à la dysfonction de la voie nigrostriée dopaminergique. Il comprend la triade akinésie, tremblement de repos, et hypertonie plastique. Un dysfonctionnement à d'autres niveaux du réseau des ganglions de la base est à l'origine de différents mouvements anormaux, chacun avec des caractéristiques cliniques spécifiques, détaillés dans le tableau 8.4. Les mouvements anormaux peuvent s'intégrer dans de nombreuses pathologies neurodégénératives, métaboliques, toxiques ou post-infectieuses.
Tableau 8.4.
Caractéristiques des différents mouvements anormaux.
| Tremblement |
| Contraction alternée, involontaire, des muscles agonistes et antagonistes, aboutissant à une oscillation rythmique involontaire d'un segment du corps autour de son axe |
| Survenue au repos (typique du tremblement parkinsonien), à la posture (tremblement essentiel ou iatrogène) ou au mouvement dirigé vers une cible (tremblement cérébelleux) |
| Myoclonie |
| Contraction brève, brusque, involontaire, d'un muscle ou d'un groupe de muscles |
| Le plus souvent de fréquence irrégulière (non rythmique), parfois déclenchée par le mouvement ou les stimulus |
| Dystonie |
| Contraction simultanée, involontaire, des muscles agonistes et antagonistes, aboutissant à des mouvements de torsion ou à des postures anormales ; mouvements lents, parfois répétitifs |
| Survenue spontanée ou à la réalisation de certains mouvements ou postures |
| Athétose |
| Mouvement involontaire lent, reptatoire de la distalité des membres |
| Rarement isolée (lésions cérébrales néonatales) |
| Chorée |
| Mouvements involontaires brusques, non rythmiques, imprévisibles (non stéréotypés), anarchiques et aléatoires |
| Touche préférentiellement les segments de membres distaux et la face |
| Ballisme |
| Mouvements involontaires brusques de grande amplitude intéressant la partie proximale des membres ; membre projeté en avant et en dehors avec une tendance à l'enroulement et la flexion |
| Souvent lié à une lésion du noyau subthalamique controlatéral |
| Choréoathétose |
| Combine chorée et athétose |
| Étiologies nombreuses (génétiques, métaboliques, toxiques) |
| Tics |
| Mouvements brefs, rapides, souvent explosifs, stéréotypés, de survenue soudaine, fréquemment par salves |
| Ressentis comme irrépressibles mais pouvant être supprimés pendant un temps variable par la volonté |
Syndrome parkinsonien : sémiologie d'une atteinte de la voie nigrostriée dopaminergique
Le syndrome parkinsonien est le plus fréquent des syndromes extrapyramidaux. Il se caractérise par l'association de trois signes majeurs appelée triade parkinsonienne : akinésie, rigidité et tremblement de repos.
La cause la plus fréquente de syndrome parkinsonien est la maladie de Parkinson, dans laquelle la manifestation de signes de la triade est unilatérale en début de maladie et cette asymétrie persiste tout au long de la maladie. Dans les syndromes parkinsoniens d'autre étiologie (iatrogène, vasculaire, post-traumatique, infectieux, autres affections dégénératives), les signes sont habituellement bilatéraux et symétriques et souvent associés à d'autres signes neurologiques.
Akinésie
Elle est définie par un ralentissement à l'initiation d'un mouvement. Il s'agit du signe caractéristique et essentiel du syndrome parkinsonien. Elle traduit la perte de mouvements automatiques, expliquant la rareté et la difficulté de mise en route du geste, qui devient lent et difficile, parfois impossible. L'akinésie gêne la réalisation des gestes de la vie quotidienne (se raser, se boutonner, se brosser les dents, couper les aliments, marcher, etc.). Elle s'associe presque constamment à une bradykinésie (diminution de la vitesse d'un mouvement) et à une hypokinésie (diminution de l'amplitude d'un mouvement), relevées à l'examen lors de la réalisation des gestes alternatifs rapides (opposition pouce-index, épreuve de marionnettes, battre la mesure avec le pied). L'akinésie est pour une grande part à l'origine de l'amimie faciale du patient (perte de mouvements automatiques de la face lors des émotions ou de la parole, rareté du clignement) et d'une pauvreté globale de mouvements spontanés. L'akinésie se manifeste également par des troubles de la marche : le démarrage devient difficile, « bégayant "(abasie trépidante ou freezing), la marche est ralentie, à petits pas, avec un polygone de sustentation étroit et une réduction ou perte du ballant des bras. Parfois le patient a une tendance à hâter le pas (démarche festinante). Les émotions, la fatigue, un obstacle visuel peuvent déclencher ou aggraver l'akinésie. De plus, ce signe moteur peut varier dans le temps (fluctuations circadiennes). Enfin, des « kinésies paradoxales » peuvent survenir lors d'un choc émotif : le malade reprend une activité motrice normale pendant de brefs instants.
Rigidité ou hypertonie extrapyramidale
La rigidité ou hypertonie extrapyramidale intéresse tous les groupes musculaires mais prédomine sur les muscles antigravitaires. Il s'agit d'une hypertonie plastique : elle se manifeste par une résistance continue et homogène à l'allongement du muscle, lors de la mobilisation passive d'un segment de membre (rigidité en « tuyau de plomb »). Souvent cette résistance cède par à-coups, réalisant le phénomène de « la roue dentée ». Dans sa forme extrême, le segment du membre mobilisé conserve la position qui vient de lui être donnée. L'hypertonie plastique peut être déclenchée ou accentuée au cours d'un mouvement du membre controlatéral (signe de Froment). Elle disparaît au cours du sommeil. La rigidité peut être douloureuse. Elle est à l'origine des modifications posturales et des mains observées chez les patients atteints de la maladie de Parkinson : tête et tronc penchés en avant (camptocormie, figure 8.1) ou tronc dévié d'un côté (syndrome de Pise, figure 8.2), les genoux en légère flexion et adduction, les bras également fléchis et collés au corps, les poignets en extension. Le faciès est figé, la paupière supérieure rétractée.
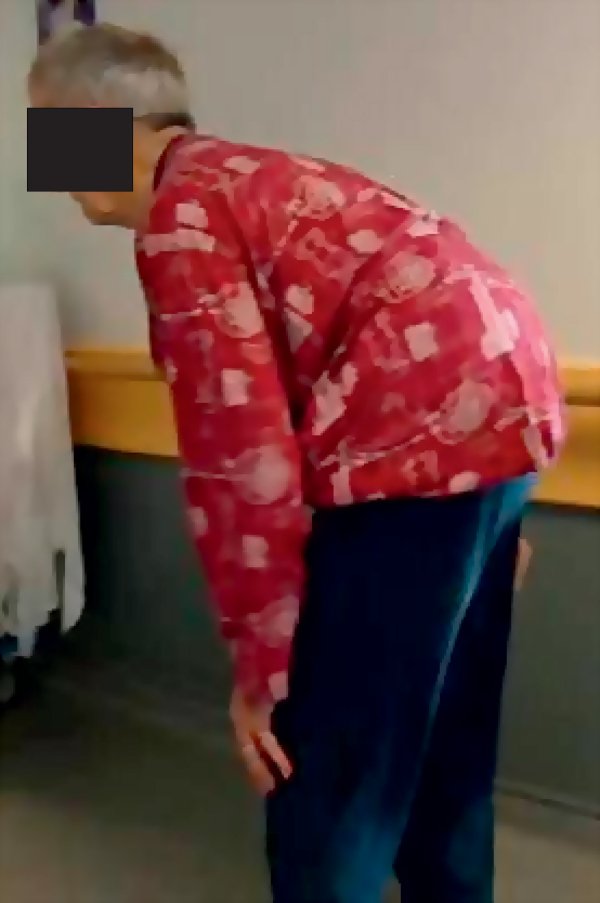
Figure 8.1
Camptocormie.
Figure 8.2
Syndrome de Pise.
Tremblement de repos ou parkinsonien
Le tremblement est défini par une oscillation rythmique involontaire de tout ou partie du corps autour de sa position d'équilibre.
Le tremblement de repos est caractérisé par des oscillations au rythme lent et régulier (4 à 7 Hz). Il se manifeste, lors du relâchement musculaire, sur un segment de membre placé en appui (mains reposant sur un accoudoir chez un patient décontracté). Les mains étant au repos lors de la marche, un tremblement d'une main survenant lors de la marche est habituellement un tremblement parkinsonien. Il disparaît, ou s'atténue considérablement, lorsque le muscle concerné se contracte pour maintenir une attitude ou exécuter un mouvement. Il prédomine aux membres supérieurs, en particulier à la main, simulant l'acte d'émietter du pain ou de rouler une cigarette. Il peut intéresser également les membres inférieurs, les lèvres ou le menton (mais pas la tête ou la voix). Il peut être unilatéral. Il est majoré par la fatigue, les émotions et le calcul mental. Il disparaît au cours du sommeil.
La triade symptomatique retentit dans d'autres domaines de la motricité, en particulier la parole et l'écriture. L'élocution est monocorde et monotone avec des troubles du débit. La parole est rare et lente, paucisyllabique (bradyphémie) ou au contraire accélérée (tachyphémie). L'écriture est caractérisée par une réduction de la taille des lettres qui s'accentue à la fin de chaque ligne et d'une ligne sur l'autre (micrographie, figure 8.3).

Figure 8.3
Exemple de micrographie.
Enfin, d'autres signes cliniques extrapyramidaux peuvent s'observer, avec une valeur diagnostique mineure, comme l'hypersialorrhée (hypersalivation) et l'hypercrinie sébacée (aspect luisant de la peau).
En fonction de la manifestation complète ou incomplète de la triade parkinsonienne, on observe plusieurs formes cliniques de syndromes parkinsoniens : forme tremblante pure (sans akinésie ou rigidité), forme akinétorigide (sans tremblement), forme triple (« complète »), forme associée à d'autres signes ou syndromes neurologiques.
Syndrome parkinsonien
- Le syndrome parkinsonien est caractérisé par l'akinésie, la rigidité et le tremblement : c'est la triade parkinsonienne qui peut être complète ou incomplète.
- Le tremblement de repos est un tremblement lent, qui disparaît ou diminue à la contraction musculaire et est augmenté par la fatigue et par l'émotion.
- L'akinésie se manifeste par une diminution de la vitesse et de l'amplitude des mouvements, une marche à petits pas, une perte du ballant automatique de l'un ou des deux bras, ainsi qu'un faciès figé.
- La rigidité correspond à une hypertonie plastique qui prédomine sur les muscles fléchisseurs et est caractérisée par un phénomène de la roue dentée à l'examen physique. Sa mise en évidence est facilitée par un mouvement controlatéral (signe de Froment).
Mouvements anormaux involontaires
- Les mouvements anormaux involontaires peuvent être rythmés et réguliers comme le tremblement (tremblements de repos parkinsonien, tremblement d'attitude et tremblement d'action, cérébelleux) ou les myoclonies rythmées.
- Les mouvements anormaux involontaires peuvent être non rythmés et lents comme dans la dystonie (« spasme ») et dans l'athétose ou être brusques et brefs comme dans les myoclonies non rythmées, la chorée et le ballisme, les dyskinésies ou les tics.
Coordination et équilibre
Thomas Wirth , Mathieu Anheim , Bruno Brochet Relecteurs : et Elisabeth Ruppert
Rappel anatomophysiologique
Situé à la partie dorsale du tronc cérébral auquel il est relié par les pédoncules cérébelleux, inférieurs, moyens et supérieurs, le cervelet intègre les informations sensorielles proprioceptives et vestibulaires ainsi que le programme moteur mis en œuvre pour coordonner finement l'action des muscles agonistes et antagonistes. Il est essentiel pour les mouvements dirigés vers une cible, les mouvements alternatifs rapides, les mouvements oculaires, et l'équilibre. Le cervelet est constitué d'une substance grise périphérique ou cortex du cervelet, d'une substance grise centrale ou noyaux cérébelleux, et d'une substance blanche (cf. chapitre 1). Les afférences (proprioception, système vestibulaire, programme moteur) se projettent sur les cellules de Purkinje dans le cortex cérébelleux. Ces cellules exercent un contrôle inhibiteur sur les noyaux cérébelleux, qui sont eux-mêmes à l'origine des efférences cérébelleuses vers le cortex moteur.
Le cervelet comprend trois zones fonctionnelles :
- le lobe flocculonodulaire à sa partie antéro-inférieure, qui reçoit des afférences vestibulaires et visuelles. Il se projette directement sur les noyaux vestibulaires dans le bulbe. Il exerce une activité de contrôle sur la musculature axiale pour maintenir l'équilibre, et sur l'oculomotricité ;
- le vermis et la partie médiane des hémisphères cérébelleux, qui reçoivent des informations proprioceptives inconscientes. Le vermis contrôle les adaptations posturales impliquant la musculature axiale et les segments proximaux des membres, tandis que la partie médiane des hémisphères cérébelleux contrôle la coordination entre muscles agonistes et antagonistes lors des mouvements volontaires ;
- la partie latérale des hémisphères cérébelleux, qui reçoit des afférences des aires associatives corticales controlatérales (cortex prémoteur, aires associatives sensorielles) et est impliquée dans la planification du mouvement juste avant qu'il soit effectué.
Corrélation anatomoclinique
Syndrome cérébelleux axial
L'ataxie cérébelleuse est la composante principale du syndrome cérébelleux. Cette phénoménologie clinique témoigne classiquement d'une atteinte du vermis cérébelleux.
Syndrome cérébelleux segmentaire
Quand la composante segmentaire prédomine, il s'agit en général de lésions affectant les hémisphères cérébelleux ou les voies cérébelleuses afférentes ou efférentes.
Lésion homolatérale ou controlatérale
Seules les lésions en regard des tractus rubrospinaux ou des noyaux ventrolatéraux thalamiques sont responsables d'un syndrome cérébelleux controlatéral à la lésion. Dans les autres cas, du fait d'une double décussation ou d'une absence de décussation, le syndrome cérébelleux est homolatéral à la lésion.
Outre le contrôle cérébelleux, d'autres structures, telles que le système vestibulaire et la sensibilité profonde consciente participent également au bon déroulement du mouvement. Une lésion à leur niveau résulte en une perturbation de la coordination et de l'équilibre avec des symptômes et signes cliniques spécifiques.
Examen clinique
Les signes cliniques résultant d'un trouble de la coordination motrice sont regroupés sous le nom d'ataxie, qui peut être statique (trouble de l'équilibre à la station debout), locomotrice (trouble de la marche) et/ou cinétique (perturbation du geste volontaire).
- Pour rechercher une ataxie statique, on demande au patient de se tenir debout pieds joints yeux ouverts, puis yeux fermés : la survenue d'oscillations du tronc indique un trouble de coordination des muscles posturaux. Le signe de Romberg correspond à l'aggravation des oscillations à la fermeture des yeux ; il est dit latéralisé quand la fermeture des yeux provoque une latéropulsion. En cas d'ataxie statique modérée, il n'y a pas d'oscillations du tronc mais seulement des contractions incessantes des tendons des muscles jambiers antérieurs (« danse des tendons »).
- L'ataxie locomotrice est définie par une marche instable, éventuellement avec un élargissement du polygone de sustentation, des embardées, un demi-tour décomposé ou une déviation latéralisée. La recherche d'une ataxie locomotrice peut être sensibilisée par l'épreuve de la marche en tandem (marche le long d'une ligne).
- Pour rechercher une ataxie cinétique, on demande au patient de poser le doigt sur une cible visuellement guidée de façon répétée, et en décubitus dorsal, de poser le talon sur le genou et ensuite de faire glisser le talon le long de la crête tibiale. L'ataxie cinétique est révélée par un crochetage au moment d'atteindre la cible et des oscillations quand le talon glisse le long de la crête tibiale.
Sémiologie
L'ataxie peut être liée à un dysfonctionnement du cervelet ou de l'un de ses systèmes afférents : le système vestibulaire et la sensibilité proprioceptive. Dans le cas d'une ataxie cérébelleuse, le trouble n'est pas compensé par les informations visuelles et l'ataxie n'est pas modifiée par la fermeture des yeux, à la différence de l'ataxie d'origine proprioceptive ou vestibulaire. Ces caractéristiques permettent de distinguer à l'examen clinique les ataxies d'origine cérébelleuse, vestibulaire et proprioceptive consciente (tableau 8.5).
Tableau 8.5.
Orientation sémiologique devant une ataxie.
| Signe de Romberg | Examen de la marche | Examen cinétique | Signes d'accompagnement | |
|---|---|---|---|---|
| Cérébelleuse | Absent : oscillations non modifiées par la fermeture des yeux | Marche ébrieuse avec élargissement du polygone de sustentation et embardées, demi-tour décomposé | Crochetages à l'épreuve doigt-nez et talon-genou, non modifiés par la fermeture des yeux | Autres signes cérébelleux : dysarthrie, nystagmus, asynergie, adiadococinésie, hypotonie musculaire |
| Vestibulaire | Présent, latéralisé | Latéropulsion | Déviation latéralisée des index à la fermeture des yeux | Autres symptômes vestibulaires : vertiges, nystagmus |
| Proprioceptive | Présent, non latéralisé | Talonnante | Dysmétrie à l'épreuve doigt-nez ou talon-genou aggravée par la fermeture des yeux | Hypoesthésie épicritique ou protopathique, douleurs neuropathiques |
Syndrome cérébelleux : sémiologie d'une atteinte du contrôle du cervelet
Le syndrome cérébelleux regroupe l'ensemble des signes cliniques témoignant de la dysfonction du cervelet et/ou des voies cérébelleuses afférentes ou efférentes.
Signes fonctionnels
Il s'agit fréquemment de troubles de la marche et de l'équilibre volontiers ressentis comme une instabilité, parfois décrite comme des sensations vertigineuses.
Le patient peut également avoir des troubles de la parole avec des difficultés à articuler ou à se faire comprendre. Parfois, il peut y avoir une maladresse ou un tremblement.
Signes physiques
Ataxie cérébelleuse
L'ataxie (littéralement, le désordre) cérébelleuse regroupe les troubles de la marche et de l'équilibre associés au syndrome cérébelleux.
La marche, décrite comme ébrieuse (c.-à-d. similaire à celle d'une personne alcoolisée) est instable avec des embardées en tous sens. Le patient compense éventuellement les troubles de l'équilibre en élargissant son polygone de sustentation, c'est-à-dire en écartant les jambes, tout en effectuant volontiers une abduction des bras. Dans les formes plus légères, on peut simplement voir une instabilité lors du demi-tour ou une décomposition de celui-ci. La marche en tandem (marche sur une ligne), test particulièrement sensible, est très souvent perturbée voire impossible.
En position pieds joints, il est possible de voir au niveau de la face antérieure des chevilles des contractions transitoires et répétées des tendons des muscles jambiers appelées « danse des tendons ». La position pieds joints peut être elle-même instable, avec des oscillations du corps en tous sens. Dans les formes sévères, elle peut être impossible à tenir. Il n'y a pas d'aggravation de l'instabilité lors de la fermeture des yeux (manœuvre de Romberg négative), contrairement au cas d'une ataxie proprioceptive ou vestibulaire.
Syndrome cérébelleux segmentaire
Il affecte principalement les membres et comporte :
- l'hypermétrie : elle correspond à un trouble du contrôle (en l'occurrence une exagération) de la durée et de l'intensité d'une activation musculaire lors de la réalisation d'un mouvement dirigé vers une cible. Lors de la manœuvre dite de poursuite du doigt ou de la manœuvre talon-genou, le doigt (ou le talon) du patient dépasse sa cible (l'index de l'examinateur ou la crête tibiale) puis revient sur celle-ci dans un second temps, donnant l'impression d'un crochetage. Elle est liée à une mauvaise coordination entre les muscles agonistes et antagonistes, ces derniers ayant pour fonction d'éviter que le mouvement volontaire ne dépasse son but. Elle peut être visualisée également lors de la manœuvre de Stewart-Holmes : on demande au patient de faire une flexion de l'avant-bras sur le bras contre une résistance exercée par l'examinateur, qui relâche brusquement cette résistance. L'amplitude du mouvement de flexion de l'avant-bras sur le bras est alors excessive par rapport à la norme ;
- la dyschronométrie : il s'agit d'une altération de la gestion temporelle des paramètres du mouvement volontaire avec un retard à l'initiation et à l'arrêt du mouvement. Elle est caractérisée lors des manœuvres doigt-nez ou talon-genou par une décomposition du mouvement en plusieurs séquences successives avec un ralentissement à l'approche de la cible ;
- l'adiadococinésie : il s'agit d'une difficulté à effectuer rapidement des mouvements alternatifs successifs. Elle est à différencier de la bradykinésie et de l'hypokinésie du syndrome parkinsonien. Par opposition à ces dernières, il n'y a pas de diminution progressive de l'amplitude des mouvements ni de leur vitesse dans l'adiadococinésie. En revanche, les mouvements sont décomposés, maladroits, irréguliers avec parfois des interruptions ;
- l'asynergie : il s'agit d'une altération de la faculté d'accomplir simultanément les divers mouvements qui constituent un acte. Lorsque l'on demande au patient de s'accroupir, celui-ci ne décolle pas les talons du sol. De même, couché, les bras croisés et les jambes écartées, il ne peut se relever sans que les cuisses ne fléchissent sur le bassin, tandis que les talons s'élèvent au-dessus du plan du lit ;
- le tremblement cérébelleux : il s'agit d'un tremblement d'action, principalement intentionnel ou identifié au début de la phase cinétique, lent, parfois de grande amplitude. Il est bien mis en évidence lors de la manœuvre doigt-nez ou talon-genou, et il peut parfois s'y associer un tremblement postural.
Remarque
En fait, la plupart des symptômes cérébelleux et notamment ceux liés aux altérations spatiale et temporelle du contrôle du mouvement sont le plus souvent intriqués ou même difficiles à différencier.
Hypotonie
Elle est visible au niveau des membres supérieurs lorsque le clinicien imprime de façon alternative un mouvement de rotation passif du tronc en manipulant les épaules du patient, celui-ci étant debout. Il existe alors des mouvements des membres supérieurs anormalement amples.
Les réflexes tendineux sont par ailleurs pendulaires : après percussion du tendon patellaire, le patient étant en position assise, la jambe continue brièvement d'osciller comme un pendule du fait de l'hypotonie.
Dysarthrie
La dysarthrie cérébelleuse est un trouble de la parole caractérisé par une voix lente, scandée, brusque, impulsive et explosive, l'amplitude de la voix étant mal coordonnée.
Troubles oculomoteurs
Plusieurs troubles oculomoteurs peuvent s'observer dans le syndrome cérébelleux.
On peut observer une hypermétrie des saccades oculaires lorsque les yeux passent d'une cible visuellement guidée à une autre. Ces mouvements oculaires dépassent la cible avant d'y revenir (saccade de « refixation »).
À l'inverse, les saccades oculaires peuvent être hypométriques et s'arrêter avant d'atteindre leur but. La poursuite oculaire peut être dite saccadique, c'est-à-dire décomposée.
Un nystagmus, qui est un mouvement involontaire rythmique et conjugué des yeux, avec une phase rapide et une phase lente, peut parfois s'observer.
Syndrome cérébelleux
- Le syndrome cérébelleux statique est caractérisé par une ataxie cérébelleuse qui comporte un élargissement du polygone de sustentation, une marche ébrieuse et une danse des tendons à la station debout.
- Le syndrome cérébelleux cinétique est caractérisé par une hypermétrie (doigt-nez, talon-genou), une dysarthrie cérébelleuse (parole scandée, explosive) et une hypotonie avec des signes cliniques homolatéraux à la lésion.
Syndrome vestibulaire : sémiologie d'une atteinte des voies vestibulaires
Le syndrome vestibulaire regroupe l'ensemble des symptômes et signes cliniques résultant d'une lésion du système vestibulaire. Ce dernier comprend trois éléments :
- le vestibule, appareil récepteur situé, avec la cochlée, dans le labyrinthe. Les canaux semi-circulaires, au nombre de trois, contiennent un liquide endolymphatique, lui-même composé des otolithes dont le déplacement stimule les cellules sensorielles, à l'origine du nerf vestibulaire ;
- le nerf vestibulaire chemine avec le nerf cochléaire dans le méat acoustique interne ;
- les noyaux vestibulaires du tronc cérébral, en connexion avec le vermis cérébelleux, la moelle spinale cervicale, la substance réticulée du tronc cérébral, les noyaux oculomoteurs et le cortex cérébral.
L'appareil vestibulaire a pour fonction de maintenir l'équilibre de l'axe du corps et la stabilité oculaire pendant le mouvement.
Dans le labyrinthe et le méat acoustique interne, les structures vestibulaires et cochléaires ont des rapports anatomiques intimes, de sorte que lors d'une atteinte périphérique (labyrinthe ou nerf cochléovestibulaire), il y a le plus souvent (mais pas toujours) association de signes cochléaires et vestibulaires. Inversement, dans les atteintes centrales (noyaux et voies vestibulaires), l'atteinte est dissociée, avec seulement des signes vestibulaires (sans signe cochléaire).
Symptômes
Vertiges
C'est le maître symptôme du syndrome vestibulaire. Il est défini par une illusion de déplacement du sujet par rapport aux objets environnants ou des objets environnants par rapport au sujet. C'est typiquement une sensation de rotation « comme un manège qui tourne ». Étymologiquement, vertigo vient du latin verso, versare : tourner. Mais il peut aussi s'agir d'impressions différentes : déplacement du corps dans le plan vertical « comme dans un ascenseur », ou instabilité, décrite comme un tangage « comme sur un bateau ». Les patients décrivent parfois seulement une impression de « tête qui tourne » sans véritable illusion de déplacement, mais la notion d'un déclenchement ou d'une aggravation par les changements de position prend alors une valeur diagnostique certaine. Lors de vertige intense, il existe des signes végétatifs d'accompagnement : nausées, vomissements, pâleur, sueurs, ralentissement du pouls. Un vertige intense est particulièrement pénible et souvent angoissant.
Troubles de l'équilibre et de la marche
Un déséquilibre à la marche peut dominer la symptomatologie, le vertige stricto sensu étant au second plan ou absent. Le malade peut décrire des embardées latérales, toujours du même côté, lors de la marche. Il peut aussi ne s'agir que d'une simple instabilité. Dans les vertiges intenses, la station debout est impossible.
Signes cliniques
Nystagmus
C'est un mouvement involontaire, rythmique et conjugué des yeux, fait de deux secousses inégales, l'une lente et l'autre rapide, cette dernière définissant arbitrairement le sens du nystagmus. Le plus souvent, il n'apparaît que lors de la poursuite oculaire. Cependant, quelques secousses nystagmiques rapidement épuisables n'apparaissant que dans les regards extrêmes n'ont pas de valeur sémiologique. Le nystagmus peut être horizontal, horizontorotatoire, rotatoire, vertical ou multiple. Plus rarement, il existe dans le regard de face (nystagmus spontané ou axial) ou lors de certaines positions de la tête (nystagmus de position).
Troubles de l'équilibre et de la marche : ataxie vestibulaire
L'ataxie vestibulaire est caractérisée par des déviations toniques axiales (index, marche aveugle en étoile).
Les troubles de l'équilibre se manifestent lors de la station debout, pieds joints avec apparition d'une inclinaison latérale, lente de l'axe du corps après quelques secondes d'occlusion des yeux. Cette déviation se fait toujours dans le même sens. C'est le signe de Romberg labyrinthique, à distinguer du signe de Romberg proprioceptif. Lors de la même manœuvre, ou le patient assis mais sans appui dorsal, les bras sont tendus en avant, les index pointés face à ceux de l'examinateur. À l'occlusion des yeux, apparaît une déviation des index qui se fait dans un plan horizontal, du même côté que le signe de Romberg. Dans les syndromes vestibulaires intenses, la station debout est impossible.
Les troubles de la marche sont caractérisés par une démarche de type ébrieux, marquée par des pulsions latérales, ou embardées. La marche aveugle consiste en la réalisation de trois pas en avant et trois pas en arrière, les yeux fermés, elle peut se faire « en étoile ». Dans les syndromes vestibulaires intenses, la marche est bien évidemment impossible, comme la station debout.
Syndromes vestibulaires périphérique et central
On distingue un syndrome vestibulaire périphérique consécutif à une atteinte du labyrinthe ou du nerf cochléovestibulaire, d'un syndrome vestibulaire central, en lien avec une atteinte des noyaux vestibulaires du tronc cérébral richement connectés.
- Dans le syndrome vestibulaire périphérique, on observe une grande crise de vertige rotatoire, clouant le malade au lit, avec vomissements et anxiété. La sémiologie cochléaire est souvent présente avec des acouphènes décrits par des bourdonnements d'oreilles et une hypoacousie. Le signe de Romberg labyrinthique, la déviation des index, les latéropulsions à la marche, se font du côté lésé, du même côté que la secousse lente du nystagmus. Il existe une composante horizontorotatoire au nystagmus dans le cadre d'une atteinte vestibulaire périphérique. C'est pourquoi le syndrome vestibulaire est dit « harmonieux ».
- Dans le syndrome vestibulaire central, les troubles de l'équilibre sont au premier plan par rapport aux vertiges, modérés ou absents. Il y a cependant des exceptions, et certains syndromes vestibulaires centraux comportent un grand vertige. En contraste avec la discrétion du vertige, le nystagmus est volontiers franc, et parfois localisateur : rotatoire (lésion bulbaire) ou vertical (lésion mésencéphalique). Les autres anomalies d'examen clinique, le signe de Romberg et la déviation des index sont absentes ou non systématisées. Le syndrome est dit « dysharmonieux ». Il n'existe habituellement pas de signes cochléaires mais fréquemment des signes neurologiques associés d'atteinte du tronc cérébral.
- Le vertige et le nystagmus peuvent être reproduits par injection d'eau froide ou d'eau chaude dans les oreilles (épreuves vestibulaires calorimétriques d'interprétation délicate).
Attention
Cette distinction entre syndrome vestibulaire périphérique et central est souvent difficile, et le meilleur élément d'orientation étiologique est la durée du vertige et ses circonstances d'apparition.
Syndrome vestibulaire
- Dans le syndrome vestibulaire, le vertige correspond à une illusion de déplacement, typiquement rotatoire et peut être accompagné de nausées, de vomissements ou d'une anxiété.
- Le nystagmus n'apparaît le plus souvent que lors de la poursuite oculaire.
- L'ataxie vestibulaire est caractérisée par un signe de Romberg labyrinthique, une déviation des index et une latéropulsion à la marche. Au maximum, la station debout est impossible.
- Le syndrome vestibulaire périphérique correspond à d'intenses vertiges accompagnés de signes cochléaires (acouphènes, hypoacousie) et réalise un syndrome « harmonieux ».
- Le syndrome vestibulaire central correspond davantage à une instabilité qu'à un vertige, avec absence de signes cochléaires mais présence fréquente d'autres signes neurologiques. Le syndrome est dysharmonieux, le nystagmus est évident.
Ataxie proprioceptive : sémiologie d'une atteinte des voies proprioceptives conscientes
L'ataxie proprioceptive est caractérisée par une atteinte de la sensibilité profonde consciente.
La marche du patient talonnante : il lance brusquement la jambe et pose le pied au sol par le talon, il « ne sait plus » où se situe son pied dans l'espace par rapport au sol avec une mauvaise perception de celui-ci. Parfois, on observe un élargissement du polygone de sustentation. Les troubles de la marche et de l'équilibre s'aggravent à la fermeture des yeux et dans l'obscurité. La manœuvre de Romberg est positive avec une chute brutale immédiate non latéralisée lors de la fermeture des yeux. C'est le signe de Romberg proprioceptif ou sensitif. Il peut s'y associer des troubles sensitifs subjectifs et des troubles sensitifs profonds objectifs. Le sens vibratoire ou pallesthésie, exploré à l'aide d'un diapason posé sur les surfaces osseuses sous-cutanées, est altéré. Il peut y avoir une altération du sens de position d'un segment de membre, le malade ayant les yeux fermés. On recherche des erreurs au sens de position du gros orteil ou du pouce, lors de l'épreuve de préhension aveugle du pouce. On peut observer une brusquerie du geste volontaire lors des épreuves doigt-nez, talon-genou avec une mauvaise direction du geste compensée par des manœuvres de rattrapage sous contrôle de la vue et aggravation des troubles lors de la fermeture des yeux. Un déficit proprioceptif peut être accompagné d'une pseudo-athétose, des mouvements involontaires qui ressemblent à l'athétose, mais disparaissent lorsque le segment de membre repose sur un plan, qui supprime la pesanteur. Lors de la « main instable ataxique, les doigts sont animés de mouvements reptatoires, aggravés par l'occlusion des yeux, mais disparaissant ou très atténués lorsque la main repose sur un plan.
Dans l'ataxie proprioceptive, il n'y a pas de vertige, de dysarthrie, d'hypotonie ni de nystagmus.
Ataxie proprioceptive
L'ataxie proprioceptive est caractérisée par une marche talonnante, une manœuvre de Romberg positive (une chute brutale non latéralisée à la fermeture des yeux), une altération du sens de position d'un membre et une hypo ou apallesthésie
Ataxie frontale : sémiologie motrice d'une atteinte des régions préfrontales, bilatérales, corticales ou sous-corticales
L'ataxie frontale est associée à d'autres signes cliniques frontaux précisés dans la sémiologie des fonctions cognitives (cf. chapitre 10).
Elle se manifeste surtout par des rétropulsions. La tendance à la chute en arrière est habituellement aggravée par la fermeture des yeux, spontanée ou provoquée. Cette rétropulsion peut se manifester dès la position assise. La marche est hésitante, incoordonnée, se faisant les pieds collés au sol ou comme si le patient marchait dans un marécage ; ces troubles sont parfois appelés apraxie de la marche. La démarche est habituellement très améliorée par le simple accompagnement sans soutien du malade. Au maximum, on peut observer une véritable astasie-abasie, c'est-à-dire qu'en l'absence de tout déficit moteur, le patient est incapable de se tenir debout et de marcher. Il n'y a pas d'ataxie cinétique.
Ataxie frontale
- Les troubles de la marche d'origine frontale sont caractérisés par une rétropulsion avec tendance à la chute en arrière, une marche hésitante, incoordonnée, les pieds collés au sol et améliorés par l'accompagnement simple.
- L'astasie-abasie correspond à une incapacité à se tenir debout et à marcher en l'absence de tout déficit moteur.
Voir QRM chapitre 32.