Le CEN vous met à l’epreuve !
Testez vos connaissances avec le quizz !
Une cause d’ataxie subaigüe à laquelle il faut penser
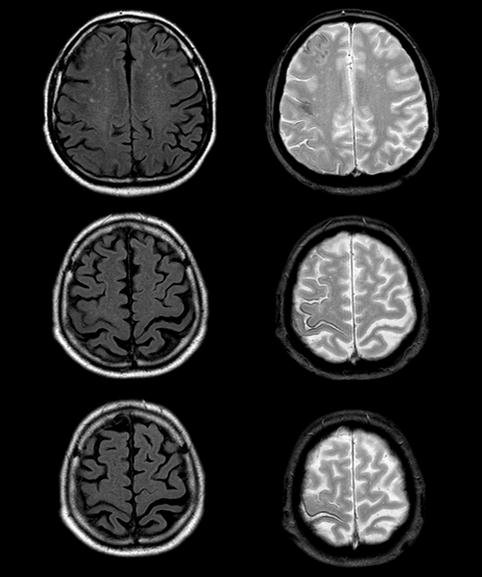
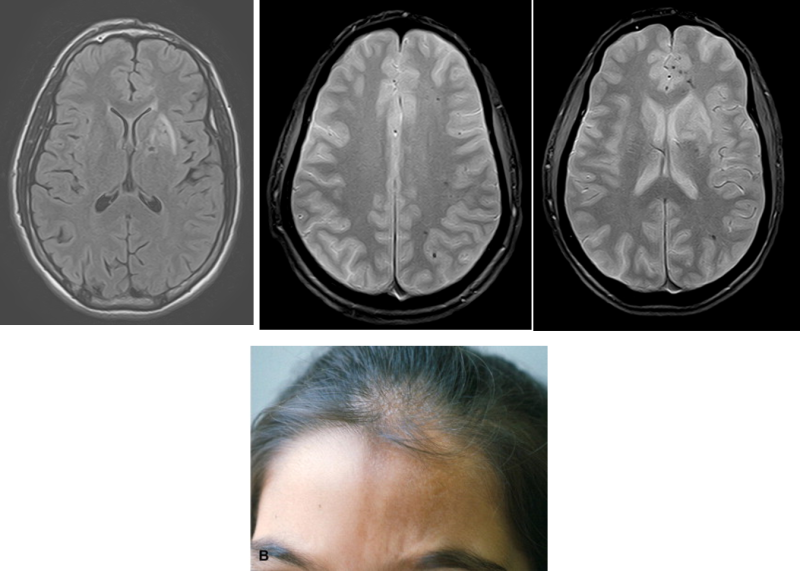
Une femme âgée de 66 ans, se présente à votre consultation pour des troubles de l’équilibre évoluant depuis 3 semaines.
L’interrogatoire retrouve des paresthésies quadri-distales évoluant depuis 1 mois, et une instabilité à la marche majorée dans l’obscurité. L’examen neurologique note un signe de Babinski bilatéral, des ROT achilléens abolis, une hypopallesthésie des 2 membres inférieurs et l’absence de déficit moteur.
La patiente rapporte dans ses antécédents un diabète de type 2 traité par Metformine depuis 3 ans et une thyroïdite d’Hashimoto substituée correctement par Lévothyroxine. Son dernier bilan biologique de routine retrouve uniquement une discrète anémie 11 g/dl avec un VGM à 108 fL.
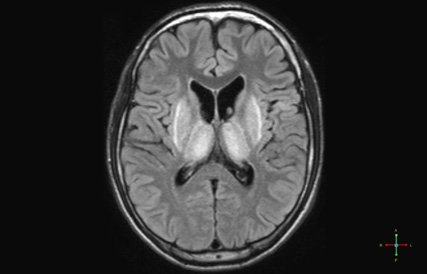
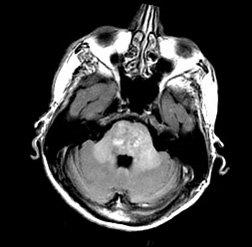
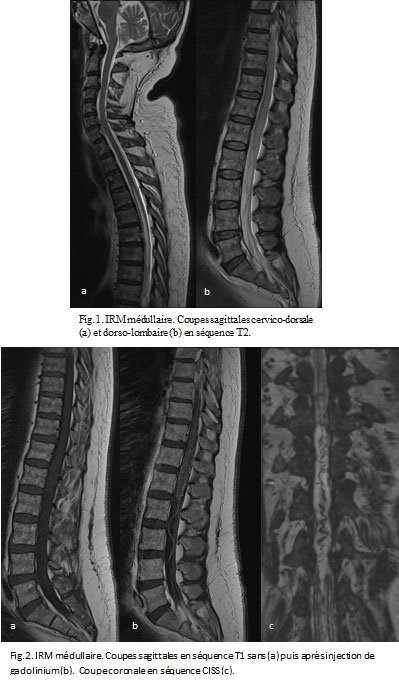
Devant ce tableau clinique vous demandez une IRM médullaire.
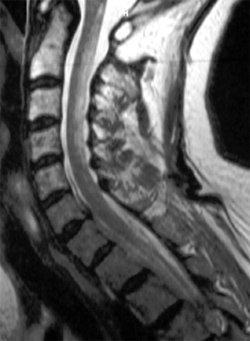
- Comment l'interprétez-vous ?
- Devant l'ensemble de ces éléments, qurel diagnostic évoquez-vous ?
Une sclérose combinée de la moelle, par probable carence en vitamine B12 devant :
- la présence d’une ataxie proprioceptive associée à des signes d’irritation pyramidale d’installation subaigüe en quelques semaines
- la mise en évidence à l’IRM d’un hypersignal en T2 au niveau des cordons postérieurs, touchant plusieurs myelomères (plutôt localisé à 1 ou 2 niveaux en cas de SEP)
- les antécédents et les traitements de la patiente qui peuvent suggérer la présence d’une carence en vitamine B12 :
- fréquente sous Metformine
- cause possible d’une anémie macrocytaire
- le terrain auto immun qui doit faire rechercher une maladie de Biermer par dosage du taux de gastrinémie, des AC anti facteur intrinsèque et anti cellules pariétales puis réalisation d’une FOGD à la recherche d’une gastrite atrophique.
Dans ce contexte, un dosage du taux sérique de vitamine B12 et d’homocystéine doit être réalisé et un traitement d’épreuve par injection IM de vitamine B12 doit être réalisé avant l’obtention des résultats des anticorps.
Du rire aux larmes
Vous voyez en consultation Mme G. âgée de 49 ans amenée par son époux pour des troubles psycho-comportementaux.
Elle n’a pas d’antécédent personnel. Vous notez une fin de vie difficile pour son père avec troubles psycho comportementaux alors qu’il était en fauteuil roulant (on aurait parlé de la maladie de Charcot). Son frère plus âgé aurait des difficultés à marcher et serait en cours de bilan neurologique.
Mme G. vous regarde avec un grand sourire et vous explique que tout va bien. Son mari raconte qu’il vit un enfer depuis plusieurs mois. Son épouse institutrice en maternelle n’arrive plus à gérer sa classe. Elle ne parvient pas à organiser l’enseignement et à même frappé des enfants si bien que des demandes de confrontation sont en cours avec des parents. A la maison, le mari note de nombreux oublis, des passages de coq à l’âne, une impatience importante nouvelle (elle ne peut plus coudre ou lire car se désintéresse vite). Ses amis se sont éloignés car elle est devenue plus franche avec de nombreuses remarques déplacées. Le mari note aussi une négligence physique puisqu’elle ne se maquille plus et ne change que très rarement ses habits. Il décrit aussi une prise de 10 kg sur un an car elle mange sans cesse des gâteaux alors qu’elle mangeait peu de sucré par le passé. L’autonomie est grandement altérée.
L’examen clinique ne retrouve pas d’anomalie.
L’examen neuropsychologique retrouve un syndrome dysexécutif comportemental et cognitif, un ludisme important et beaucoup de triche durant les passations. La mémoire est conservée, il n’y a pas de trouble instrumental. Elle n’a malheureusement pas pu être explorée sur le plan orthophonique.
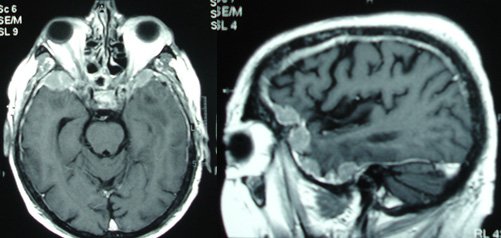
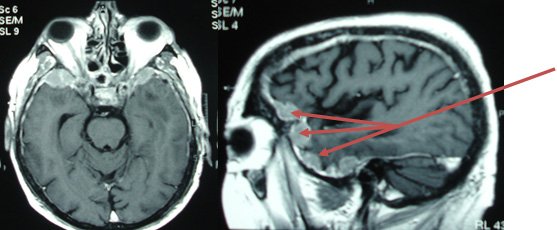
Voici une IRM cérébrale et un PET-Scanner.
Que dire de l’imagerie ? Quel est le diagnostic neuropsychologique ? Quel diagnostic plus général peut-on proposer ?
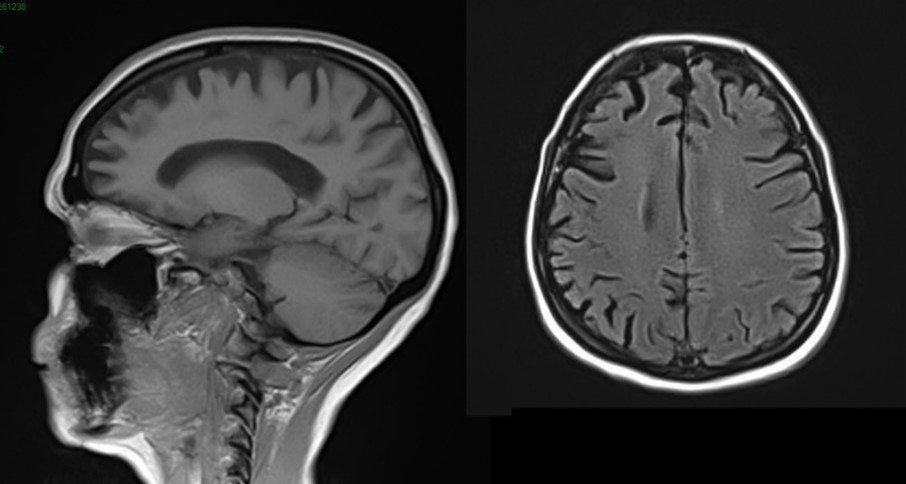
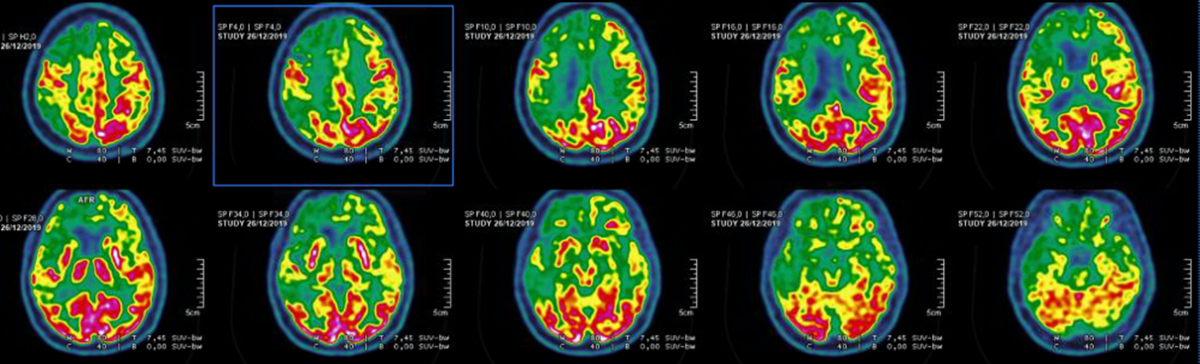
Atrophie frontale sur l’IRM avec hypométabolisme antérieur marqué, démence fronto-temporale, tableau de mutation C9ORF72
La démence frontotemporale (DFT) fait partie du groupe des dégénérescences lobaires fronto temporales (DFT, DCB, PSP, APP non fluente et sémantique). Ici, le tableau clinique remplit les critères cliniques de DFT possible selon Rascovsky, soit au moins 3 critères parmi :
Désinhibition comportementale précoce,
Apathie/inertie précoce,
Perte de sympathie ou d’empathie précoce,
Comportement persévératif, stéréotypé ou compulsif/obsessionnel précoce,
Hyperoralité et changement des habitudes alimentaires,
Profil neuropsychologique (déficit exécutif, préservation mémoire épisodique et fonctions visuo spatiales).
Le diagnostic devient probable car aux critères cliniques de DFT possible s’ajoute un déclin fonctionnel significatif et des résultats d’imageries concordants (IRM et PET).
L’histoire familiale permettra probablement de passer en stade certain puisqu’elle semble orienter vers une mutation C9ORF72.
On estime à 40% l’origine génétique des DFT. Cela motive la recherche systématique d’une histoire familiale. La mutation C9ORF72 est finalement de description récente mais probablement la plus fréquente. Elle doit systématiquement être évoquée dans une histoire familiale mêlant DFT et SLA.
Bibliographie :
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. >Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain [Internet] 2011;134(9):2456–2477
De Jesus-Hernandez M.Mackenzie IR, Boeve BF et al.Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72(2):245-56.
Renton AE, Majounie E, Waite A et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72(2):257-68.
Une histoire renversante
Vous voyez en consultation un patient âgé de 66 ans. Il a comme antécédents une agression physique en 2003 avec un état de stress post traumatique séquellaire et une toux chronique depuis 10 ans avec bilan étiologique pneumologique et gastro-entérologique négatif.
Depuis l’agression, il décrit l’installation progressive de troubles de la marche avec chutes itératives parfois traumatiques.
Votre examen clinique retrouve une marche ataxique avec élargissement du polygone de sustentation, un Romberg positif, une marche en funambule impossible et aucun déficit moteur. Les réflexes tendineux ne sont pas perçus. La pallesthésie est abolie aux membres inférieurs jusqu’aux genoux et aux membres supérieurs jusqu’aux coudes.
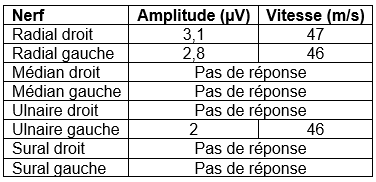
L’étude des vitesses de conduction motrices est normale comme l’examen de détection à l’aiguille. L’étude des vitesses de conduction sensitives vous est proposé ci-dessous. Il n’y a aucune réponse reproductible aux quatre membres lors de l’enregistrement des potentiels évoqués somesthésiques. Une IRM cérébrale est réalisée. Une coupe sagittale pondérée en T1 est proposée ci-dessous.
Quel est votre diagnostic ?
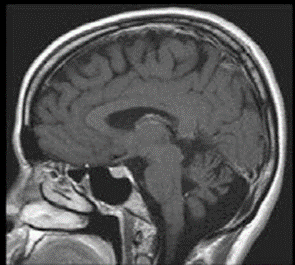
Syndrome de CANVAS
Il s’agit d’un syndrome de CANVAS (Cerebellar Ataxia Neuropathy Vestibular Areflexia Syndrome) caractérisé par la triade trouble cérébelleux, atteinte vestibulaire bilatérale et déficit sensitif. L’atteinte neurologique est d’évolution lente et d’apparition tardive à l’âge adulte. On peut également retrouver dans presque deux-tiers des cas une toux chronique et dans un tiers des cas des symptômes dysautonomiques.
En général les potentiels évoqués somesthésiques sont absents et l’ENMG montre un aspect de ganglionopathie. L’IRM encéphalique retrouve une atrophie cérébelleuse et l’examen vestibulaire une anomalie du réflexe visio-oculo-vestibulaire (aréflexie vestibulaire bilatérale assez caractéristique).
L’apparition d’un syndrome de CANVAS est souvent tardive et sporadique. Le diagnostic nécessite d’éliminer les diagnostics différentiels tel que le SCA3 ou la maladie de Friedreich. Des cas familiaux ont été décrit, faisant découvrir une répétition intronique AAGGG récessive dans le gène RFC1.
Bibliographie :
Szmulewicz DJ, McLean CA, MacDougall HG, Roberts L, Storey E, Halmagyi GM. CANVAS an update: clinical presentation, investigation and management. J Vestib Res Equilib Orientat. 2014;24(56):465‑74.
Szmulewicz DJ, Roberts L, McLean CA, MacDougall HG, Halmagyi GM, Storey E. Proposed diagnostic criteria for cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Neurol Clin Pract. févr 2016;6(1):61‑8.
Peillet C, Buch D, Nifle C, Servan J, Pico F. Ataxie cérébelleuse avec neuropathie et aréflexie vestibulaire bilatérale : le syndrome CANVAS, démarche diagnostique. Prat Neurol - FMC. déc 2018;9(4):268‑71.
Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yau WY, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet. avr 2019;51(4):649‑58.
Cortese A, Reilly MM, Houlden H. RFC1 CANVAS / Spectrum Disorder. 2020
Nov 25. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A, editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993–2021. PMID: 33237689.
N'oubliez pas mon IRM !
En mai 2011, sans facteur déclenchant particulier, une femme de 59 ans présenta un tableau d’amnésie pour lequel elle fut adressée au service d’accueil et des urgences. A l’admission on observait une amnésie antérograde massive, avec oubli à mesure, qui s’estompa dans l’heure suivant l’arrivée de la patiente. Durant cet épisode d’amnésie la patiente n’était pas anxieuse, ne reposait pas de façon spontanée les mêmes questions. L’examen neurologique était par ailleurs normal.
La durée totale estimée de l’épisode d’amnésie était de 2 heures. La patiente dit se souvenir du début de celui-ci, d’une sensation de malaise initial, du contexte de survenue (le matin peu après le réveil). Elle garda une amnésie lacunaire complète du reste de l’épisode.
Elle n’avait aucun antécédent médico-chirurgical. Elle dit en particulier n’avoir jamais fait d’épisode similaire. Un EEG réalisé après résolution de l’amnésie fut parfaitement normal. Les fonctions cognitives évaluées à l’aide de la MoCA étaient intègres (score de 30/30). Une IRM cérébrale fut demandée mais réalisée seulement une semaine plus tard.
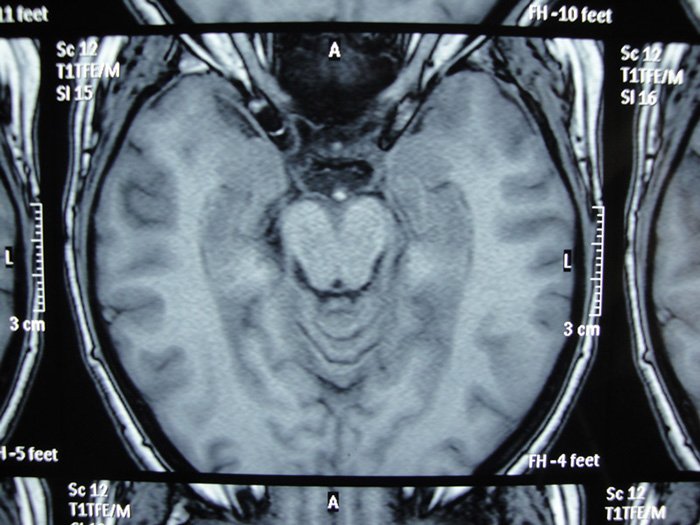
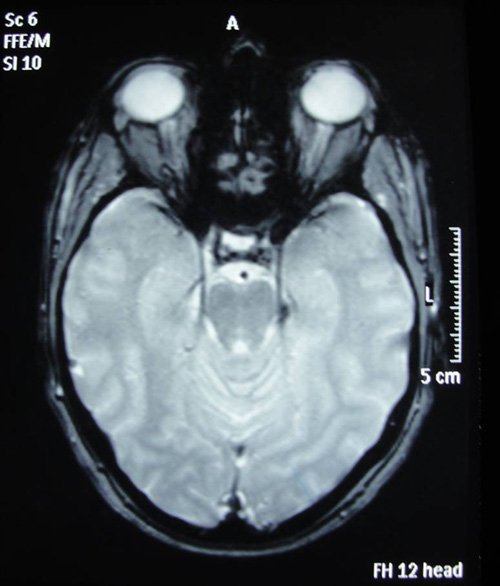
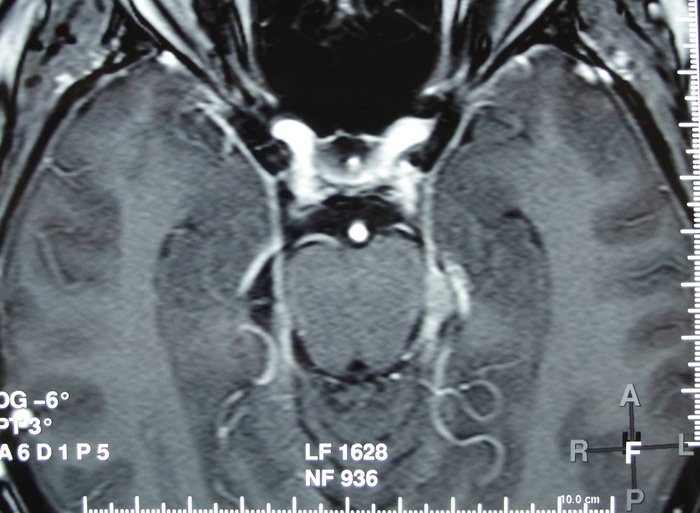
Quel-est votre diagnostic ?
Cet épisode d’amnésie antérograde de deux heures pouvait évoquer un ictus amnésique, puisqu’il réalise un tableau d’amnésie antérograde de moins de 24 heures, isolé et spontanément résolutif. La durée en était un peu brève, car la durée moyenne des ictus amnésiques s’est révélée être, sur une série de 788 cas, de 6,7 heures (Caplan, 1990) ; il n’y avait pas de facteur déclenchant tel qu’une émotion, une douleur, une exposition au chaud ou au froid, un traumatisme mineur, mais on sait que certains ictus amnésiques surviennent sans facteur déclenchant évident ; la patiente n’avait pas de terrain prédisposant, tel qu’une histoire d’anxiété, de migraine ou des facteurs de risque vasculaire, même si ce terrain peut être aussi absent dans les ictus amnésiques (Bartsch et Deuschl, 2010). Les deux éléments sémiologiques les plus atypiques étaient le mode de début, par un malaise, et le souvenir que la patiente gardait de celui-ci, puisque les ictus amnésiques s’accompagnent toujours d’un léger débord rétrograde (oubli de quelques heures à quelques jours). Il en découle que le début de l’ictus n’est jamais rappelé, alors que la fin de l’amnésie est souvent progressive (Guillery-Girard et al, 2004).
L’IRM cérébrale de haute résolution, quand elle est réalisée dans les 48 à 72 heures après un ictus amnésique, peut objectiver des lésions de 1 à 5 mm en séquences de diffusion ou en T2, interprétées comme la conséquence d’un œdème cytotoxique (Bartsch et Deuschl, 2010).
L’IRM réalisée dans notre cas dans un créneau horaire plus lâche, est venue plutôt appuyer l’hypothèse d’une amnésie transitoire symptomatique. En effet on mit en évidence une petite masse extra-axiale, oblongue, de l’échancrure tentorielle gauche, d’environ 8 mm de grand axe sagittal et de 3-4 mm dans le plan coronal. Cette masse était hypo-intense en T2 écho de gradient, en faveur d’une calcification de celle-ci et rehaussée de façon homogène et modérée par l’injection de gadolinium, Elle arrivait au contact de l’artère cérébrale postérieure gauche et était située en regard de la tête de l’hippocampe. Elle présentait toutes les caractéristiques d’un méningiome de la tente du cervelet, un peu protrusif vers la citerne ambiante, susceptible d’être épileptogène.
L’origine épileptique de l’amnésie apparaît bien plus probable. On ne peut cependant affirmer avec certitude le diagnostic d’amnésie épileptique transitoire que si une des 3 conditions suivantes est remplie : i) si les épisodes se renouvellent et disparaissent sous traitement anti-épileptique ; ii) si l’EEG montre des anomalies épileptiformes focales temporales ; iii) ou si l’amnésie est accompagnée de signes évocateurs d’une crise mésio-temporale (automatismes oro-alimentaires, hallucinations olfactives…). La sensation de malaise décrite par notre patiente au début de l’épisode, même si elle n’est pas d’une parfaite spécificité, correspondait sans doute aux premiers symptômes d’une crise mésio-temporale (Butler et al, 2007 ; Bartsch et Deuschl, 2010). L’horaire de survenue, le matin au réveil, est également plus suggestif d’une crise d’épilepsie.
Bien que la patiente pût être considérée comme souffrant d’épilepsie (prédisposition persistante à faire une crise – du fait du méningiome - et survenue d’une crise épileptique), en l’absence de récidive des épisodes amnésiques une abstention thérapeutique fut proposée.
Auteurs :
François Sellal (1), Benjamin Cretin (1), Hélène Oesterlé (2).
(1) CMRR de Strasbourg-Colmar.
(2) Service de Neuroradiologie diagnostique et interventionnelle, Hôpital Pasteur, Colmar.
Références :
- Caplan LR. Transient Global Amnesia : characteristic features and overview. In: HJ Markowitsch (ed). Transient Global Amnesia and related disorders. Toronto: Hogrefe et Huber, 1990; 15-27.
- Guillery-Girard B, Desgranges B, Urban C, Piolino C, de la Sayette V, Eustache F. The dynamic time-course of memory recovery in transient global amnesia. J Neurol Neurosurg Psychiatry 2004; 75: 1532-40.
- Bartsch T, Deuschl G. Transient global amnesia: functional anatomy and clinical implications. Lancet Neurology 2010;9: 205-14.
- Butler CR, Graham KS, Hodges JR et al. The syndrome of transient epileptic amnesia. Ann Neurol 2007;61: 587-98.
Un cerveau difficile à décrypter
Une patient de 56 ans, fut adressée aux urgences pour des vertiges et des troubles de l’équilibre responsables de chutes à répétition depuis 3 semaines. Dans ses antécédents on retrouvait des facteurs de risque cardio-vasculaires (Diabète de type 2, Hypertension artérielle, dyslipidémie, obésité), une auto-immunité (hypothyroïdie, cirrhose avec anticorps anti-muscles lisses), une ACFA et un carcinome hépatocellulaire (nodule de 22mm de découverte récente et qui aurait du être traité par radiofréquence la semaine suivante).
L’examen clinique retrouvait un syndrome cérébelleux stato-cinétique, un opsomyoclonus et un syndrome méningé.
L’IRM cérébrale retrouvait les anomalies suivantes :
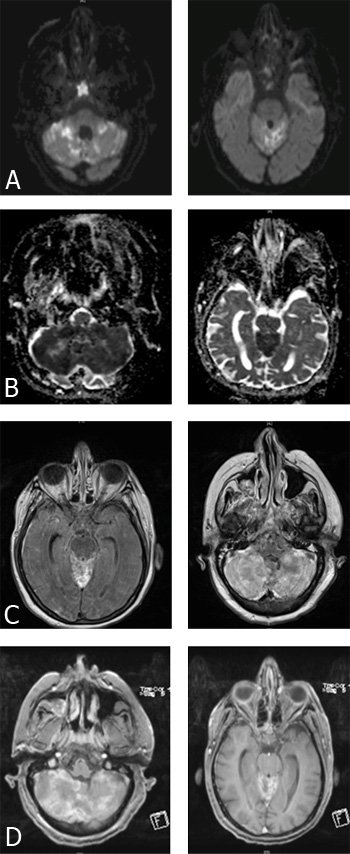
A. DIFFUSION - B. ADC - C. FLAIR - D. T1 gadolinium
La ponction lombaire montrait une hyperleucocytose à 97 cellules/mm3 à prédominance lymphocytaire, une hyperprotéinorachie à 2,63 g/l, une hypoglycorachie à 0,17 g/L, la présence de bandes oligoclonales et l’absence de germe à l’examen direct et de cellules suspectes de malignité.
- Que suspectez-vous ?
- Quels examens complémentaires proposez-vous ?
L’IRM cérébrale montre un hypersignal en diffusion et en FLAIR au niveau des deux hémisphères cérébelleux essentiellement au niveau du cortex et du vermis prenant partiellement le contraste. Les plages d’hypersignal en ADC vont à l’encontre de l’hypothèse d’un AVC. Ces imageries évoquent une cérébellite d’origine infectieuse ou paranéoplasique.
La ponction lombaire oriente vers une origine infectieuse (tuberculose, listériose, cryptococcose).
Il est nécessaire de compléter les examens par des recherches mycologiques et bactériologiques (examen direct et culture dans le LCR, recherches d’anticorps et d’antigènes anti-cryptococciques, hémocultures à la recherche de Listeria monocytogène) dans le sang et le LCR et de biopsier si possible le cervelet.
Il est également indispensable de rechercher une immunodépression cellulaire avec un typage lymphocytaire, le complément et les sérologies VIH1/2.
Evolution :
Les antigènes anti-cryptococciques, les anticorps et les cultures cryptococciques dans le sang et le LCR sont revenus positifs et la biopsie exérèse du cervelet a permis de retrouver des cryptocoques. Le reste des recherches est revenu négatif. Le typage lymphocytaire et le complément étaient sans particularité et les sérologies sont revenues négatives
Le cryptococcus neoformans est une levure ubiquitaire retrouvée dans les sols riches en fientes d’oiseaux. Il est responsable de la cryptococcose qui est une maladie opportuniste favorisée par l’immunodépression cellulaire (VIH++, chimiothérapie, corticothérapie prolongée, …) et est exceptionnelle en cas d’immunocompétence. Il a un tropisme pour le système nerveux central et est responsable de tableaux de méningo-encéphalites, myélites, cryptococcomes. En cas d’immunocompétence, il est plutôt responsable de tableaux de méningites chroniques du fait d’un système immunitaire efficace qui limite l’infection aux méninges.
La contamination se fait par inhalation via une porte d’entrée pulmonaire.
Le traitement recommandé est l’Amphotéricine B (1mg/kg) et la flucytosine (100mg/j) pendant 2 semaines puis un relai par fluconazole 400mg/j de manière prolongée. En l’absence de traitement l’infection est toujours mortelle et le taux de mortalité est de 20% malgré un traitement efficace.
Le cas de cette patiente est atypique du fait d’une atteinte cérébelleuse rare dans la cryptococcose et d’une absence d’immunodépression.
Cas proposé par : Cécilia Alves Do Rego, Strasbourg.
Pour en savoir plus :
- Neuville S et al. Physiopathologie des méningo-encéphalites dues à Cryptococcus neoformans. Ann Med Interne. 2002;5:323–8.
- Day et al.Combination antifungal therapy for cryptococcal meningitis.New England Journal of Medicine. 368;14, 2013).
- E.Pilly, 22eme edition CMIT, pages 452/453.
PSP? ... pas vraiment....
Une femme de 55 ans présentait depuis deux 9 ans des troubles de l'équilibre avec sensation de tangage, des chutes en rétropulsion et une dysarthrie hypophonique. Un an après le début des symptômes apparurent des troubles de déglutition prédominant sur les liquides et une détérioration cognitive marquée par un score de 126/144 à l'échelle de Mattis. Elle présentait comme principaux antécédents une coronaropathie et plusieurs interventions chirurgicales (chirurgie de l'uretère droit, cure de hernie inguinale et ablation de kystes mammaires). Lors de la première consultation, son traitement comportait 100 mg de piribédil et 250 mg de lévodopa associée à la bensérazide. L'examen neurologique objectivait un syndrome cérébelleux cinétique et statique, un syndrome frontal, une instabilité posturale avec rétropulsion, des réflexes ostéotendineux vifs et un ralentissement des saccades oculaires. Il n'y avait pas de syndrome parkinsonien segmentaire, ni de dysautonomie.
Biologiquement, il n'y avait pas de carence en vitamines du groupe B (B1, B6, B9 et B12), ni en vitamine E, la recherche d'anticorps anti- GAD était négative. Le bilan métabolique, comprenant la chromatographie des acides aminés et l'étude pré- et post-prandiale sur 24 h de l'ammoniémie et du rapport lactate/ pyruvate ne montrait pas d'anomalies. L'EEG, l'IRM cérébrale et la ponction lombaire ne donnaient pas d'arguments en faveur d'une maladie de Creutzfeldt-Jacob.
Environ un an plus tard, apparurent de discrets mouvements choréiques et un syndrome extrapyramidal segmentaire. L'étude en biologie moléculaire des gènes IT15, ADRLP et SCA17 était négative. Dans les mois qui suivirent, s'installa progressivement un rétrocolis, une ophtalmoparésie franche de la verticalité du regard. À l'examen, il existait une nette aggravation des troubles posturaux et du syndrome frontal avec apparition d'un signe de l'applaudissement franc.
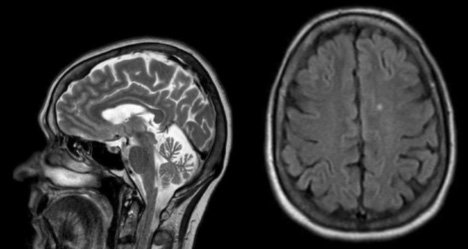
- Comment analyser-vous l'IRM ?
- Quel diagnostic évoquez-vous ?
- Quel examen complémentaire demandez-vous pour l'affirmer ?
- L’ IRM cérébrale, à gauche en séquence T2 sagittale montre une atrophie vermienne. À droite, en séquence FLAIR axiale elle montre un hypersignal non significatif de substance blanche.
- L ‘association d’une ataxie cérébelleuse, d’une ophtalmoplégie supranucléaire, d’une dysarthrie, d’une dysphagie, de troubles cognitifs, et de mouvements anormaux conduit à évoquer le diagnostic de maladie de Niemann Pick de type C
- Une mise en culture de fibroblastes cutanés pour réaliser un test à la filipine et le séquençage des gènes NPC1 et NPC2.
La maladie de Niemann Pick de type C est une pathologie du trafic intracellulaire du cholestérol de transmission autosomique récessive. Son expression est extrêmement variable en fonction de l'âge de début des symptômes. Un score prédictif a été réalisé pour aider au diagnostic (Wijburg et al). Le bilan diagnostique comporte une mise en culture de fibroblastes par biopsie cutanée pour réaliser un test à la filipine et le séquençage du gène NPC1 et NPC2. L'interprétation des tests se fait de manière combinée.
A l'âge adulte, le tableau clinique peut associer par ordre de fréquence une ataxie cérébelleuse, une ophtalmoplégie supranucléaire, une dysarthrie, des troubles cognitifs, des mouvements anormaux (syndrome parkinsonien, dystonie, chorée), des troubles psychiatriques et une dysphagie. La splénomégalie est beaucoup moins fréquente à l'âge adulte, l'épilepsie est rare.
L'âge de début est assez tardif chez cette patiente. La biopsie cutanée et la biologie moléculaire confirmèrent une maladie de Niemann Pick de type C avec une mutation hétérozygote c.1843C>T, c3019C>G.
Auteurs : G Grolez, A Dejardin, D Devos, L Defebvre, C Moreau (Lille).
Références :
Godeiro-Júnior C, Inaoka RJ, Barbosa MR, Silva MR, Aguiar Pde C, Barsottini O. Mutations in NPC1 in two Brazilian patients with Niemann-Pick disease type C and progressive supranuclear palsy-like presentation. Mov Disord. 2006;21(12):2270–2.
Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16.
Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F; NP-C Guidelines Working Group. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab. 2012;106(3):330–44.
Wijburg FA, Sedel F, Pineda M, Hendriksz CJ, Fahey M, Walterfang M, et al. Development of a suspicion index to aid diagnosis of Niemann-Pick disease type C. Neurology 2012;78 (20):1560–7 [doi: 10.1212/WNL.0b013e3182563b82. Epub 2012 Apr 18].
Sur un air d'Italie
Une patiente de 44 ans fut hospitalisée pour des crises convulsives tonico-cloniques généralisées sur la voie publique. L’examen clinique aux urgences objectivait un état cachectique, une confusion avec hémiparésie droite puis un déficit des 4 membres sans syndrome pyramidal. La marche n’était pas évaluable, il n’y a pas d’anomalie des nerfs crâniens, pas de nystagmus, ne de syndrome méningé. La patiente était apyrétique. Le bilan biologique révéla une anémie microcytaire sans syndrome inflammatoire et une élévation des gamma-GT. La recherche des toxiques était négative. Une pré-albumine effondrée et une carence en vitamines B1 et B6 étaient en faveur d’une dénutrition sévère. L’interrogatoire était impossible du fait du syndrome confusionnel. L’IRM cérébrale réalisée en urgences révéla les anomalies suivantes (Figure 1).

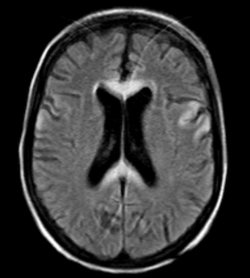
Figure 1
- Quel est votre diagnostic ?
- Quel est le mécanisme physiopathologique ?
- Le tableau clinique avec syndrome confusionnel, crises convulsives et tétraparésie associé à un hypersignal diffus du corps calleux et des hypersignaux corticaux en séquence T2 Flair (Figure 1) sont en faveur du diagnostic d’encéphalopathie de Marchiafava-Bignami.
- C’est une pathologie rare (250 cas recensé depuis 1903) dont la physiopathologie reste encore mal connue (pathologie démyelinisante et carentielle).
Figure 1
L’encéphalopathie de Marchiafava-Bignami se traduit par un syndrome confusionnel avec troubles de la vigilance, une dysarthrie, une tétraparésie avec parfois un syndrome de dysconnexion calleuse. L’imagerie reflète le processus de démyelinisation et de nécrose du corps calleux 1 puis une évolution séquellaire cavitaire (Figure 2). On peut retrouver des hypersignaux corticaux avec une probable atteinte des fibres commissurales, extra-calleuses, et des fibres associatives cortico-corticales et cortico-sous-corticales avec en anatomopathologie une nécrose laminaire corticale 2. La présence de troubles de la vigilance, d’un hypersignal diffus du corps calleux et d’hypersignaux corticaux serait de moins bon pronostic 3. Les études en spectroscopie ont montré une augmentation de la choline en phase aiguë démyélinisante et des lipides à quelques semaines du début du fait d’un envahissement macrphagique. En imagerie fonctionnelle il existe une hypoperfusion et une diminution franche du métabolisme dans les lobes frontaux et pariétaux, du gyrus orbitaire et du thalamus 4. Le bilan biologique objective souvent une carence en vitamine B1, il est difficile de savoir s’il existe un lien direct entre cette carence et les lésions du corps calleux ou si ces patients ne présentent pas également une encéphalopathie de Gayet Wernicke à bas bruit (terrain de dénutrition et/ou d’alcoolisme chronique) 5. L’évolution est toutefois plus favorable en cas de supplémentation intra-veineuse prolongé de thiamine qui reste le seul traitement ayant montré une efficacité.
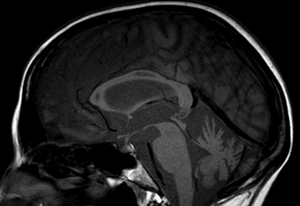
Figure 2
IRM de contrôle (séquence T1 coupe sagittale) de notre patiente à 15 jours du début des troubles avec aspect atrophié et cavitaire du corps calleux.
Proposé par Lucie Hopes (Service de Neurologie et pathologie du mouvement, CHRU de Lille).
Références :
1 Bhat A, Punia V, Lee HJ, Marks D. Corpus callosum fibre disruption in Marchiafava-Bignami dis ease. Pract Neurol. 2013.doi:10.1136/practneurol-2013-000657.
2 Johkura K, Naito M, Naka T. Cortical involvement in Marchiafava-Bignami disease. Am J Neuroradiol. 2005;26(3):670-3.
3 Heinrich A, Runge U, Khaw AV. Clinicoradiologic subtypes of Marchiafava-Bignami disease. J Neurol. 2004;251(9):1050-9.
4 Ishii K, Ikerjiri Y, Sasaki M, Kitagaki H, Mori E. Regional cerebral glucose metabolism and blood flow in a patient with Marchiafava-Bignami disease. Am J Neuroradiol. 1999;20(7):1249-51.
5 Hillbom M, Saloheimo P, Fujioka S, Wszolek ZK, Juvela S, Leone MA. Diagnosis and management of Marchiafava-Bignami disease: a review of CT/MRI confirmed cases. J Neurol Neurosurg Psychiatry. 2013 ;26.doi:10.1136/jnnp-2013-305979.
Atteinte multiple des nerfs
Un homme de 72 ans présentant dans ses antécédents un adénocarcinome prostatique métastasé aux os (en échappement thérapeutique après 4 lignes de chimiothérapie) a développé une paralysie faciale gauche périphérique depuis quelques jours dans un contexte d’altération de l’état général.
L’examen clinique objective outre cette paralysie faciale gauche, une baisse de l’acuité visuelle de l’oeil droit ainsi qu’une anisocorie avec myosis serré droit, une hypoesthésie et des dysesthésies frontales droites et une paralysie de l’abduction de l’œil droit.
La ponction lombaire réalisée retrouve : une hyperprotéinorachie à 1.48g/l et 2 GB/mm3, une glycorachie normale et l’absence de cellules anormales.
L’IRM est la suivante :
Coupes axiale et sagittale T1 injectées
Quel est votre diagnostic ?
L’IRM cérébrale injectée met en évidence :
- Prise de contraste et épaississement des leptoméninges.
- Formations micronodulaires multiples disséminées dans les méninges.
Coupes axiale et sagittale T1 injectées
Ceci traduit un envahissement des méninges par des cellules cancéreuses et une dissémination de ces cellules dans le LCR.
Il s’agit d’une méningite carcinomateuse responsable d’une atteinte multiple des nerfs crâniens par envahissement (nerfs II, V1, VI, VII droits).
Elle peut être la présentation initiale du cancer sous-jacent dans 5 à 10% des cas et est le plus souvent associée à un cancer du sein, du poumon, du rein, de l’ovaire, de la vessie, ou à un lymphome…
Les symptômes neurologiques les plus souvent retrouvés sont les atteintes multiples des nerfs crâniens, une encéphalopathie, mais également parfois une atteinte mal systématisée du système nerveux périphérique avec une composante douloureuse majeure.
L’IRM cérébro-médullaire est indispensable au diagnostic identifiant l’envahissement et l’épaississement méningé et des nerfs crâniens sous forme de prise de contraste, voire les nodules sous-arachnoïdiens ou parenchymateux.
La PL confirmera le diagnostic en retrouvant une hyperprotéinorachie dans 70% des cas, une pléiocytose dans 63% des cas et souvent une hypoglycorachie (entre 32 et 75% des cas) et en identifiant des cellules néoplasiques dans le LCR. La PL est à répéter en cas d’absence de cellules néoplasiques initialement (sensibilité de dépistage des cellules néoplasiques de 100% après la 3ème PL).
Le pronostic est très péjoratif. La médiane de survie ne dépasse pas 14 semaines malgré d’éventuels traitements par chimiothérapie intrathécale couplée à une radiothérapie plus ou moins ciblée.
Pour en savoir plus : Jayson GC, Howell A. Carcinomatous meningitis in solid tumours. Ann Oncol. 1996 (8):773-86. Gleissner B, Chamberlain MC. Neoplastic meningitis. Lancet Neurol. 2006 (5):443-52.
Troubles du comportement aigus chez une jeune femme
Une jeune femme de 19 ans, sans antécédent, présente d’installation subaigüe sur 15 jours, des troubles du comportement. On note à son arrivée aux urgences un ralentissement psychomoteur, des rires immotivés, de nombreux « coq à l’âne », l'examen somatique est sans particularité. Elle décrit de discrètes céphalées et on note un fébricule à 37.8°.
Un scanner cérébral se révèle normal. La ponction lombaire révèle une hyperlymphocytose isolée. L’ IRM cérébrale montre un hypersignal temporal en séquence FLAIR.
Le diagnostic de méningo-encéphalite herpétique est évoqué.
Un traitement par Aciclovir 10 à 15 mg/kg toutes les 8 heures en IV est entrepris en urgence.
Les résultats de la PCR du virus de l’herpès dans le LCR revient rapidement et est négative.
En raison des troubles du comportement, un EEG (figure 1 et 2) est réalisé
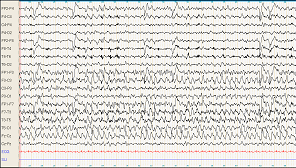
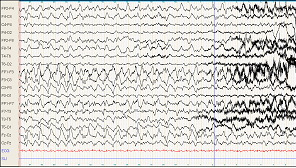
- Comment interprétez-vous cet EEG ?
- Un autre diagnostic doit-il être évoqué ?
- Quel autre examen complémentaire suggérez-vous ?
- crise partielle temporale gauche se généralisant Figure 1 : présence de signes paroxystiques avec pointes ondes temporales gauches en faveur d'une crise partielle temporale gauche. Figure 2 : présence de pointes ondes généralisées
- le diagnostic d'encéphalite limbique dysimmunitaire doit être évoqué
- la recherche d’anticorps anti neurones et notamment anti NMDA et anti VGKC doit être réalisée dans le sang et le LCR
Les encéphalites dysimmunitaires peuvent débuter sur un mode subaigu par :
- des troubles de l'humeur, une irritabilité, une agitation, une confusion, un état dépressif majeur
- des troubles mnésiques sévères atteignant principalement la mémoire antérograde, avec hallucinations, ou une démence
- des crises épileptiques avec pharmacorésistance
On peut également :
- observer des troubles de la vigilance d'apparition brutale conduisant à une hospitalisation en réanimation.
- retrouver une phase prodromique de type viral avec hyperthermie et céphalées précédant la survenue des signes neurologiques ou psychiatriques
Les examens complémentaires à réaliser sont les suivants chez une femme jeune :
- Dosage des anticorps anti neuronaux dans le sang et le LCR : notamment Ac anti NMDA (positifs chez cette patiente), et anti VGKC
- Réalisation d'un scanner thoraco abdomino pelvien à la recherche d'une néoplasie, et en complément une IRM pelvienne chez la jeune fille car la tumeur la plus fréquente est le tératome ovarien.
- L'IRM cérébrale reste souvent aspécifique, avec un hypersignal temporal en séquence FLAIR
Le traitement de la tumeur, ou en l’absence de tumeur, les traitements immunomodulateurs peuvent permettre une évolution favorable des signes neurologiques.
Cas proposé par Alice ROBBE, Angers.
Pour en savoir plus : Vitaliani R, Mason W, Ances B, Zwerdling T, Jiang Z, Dalmau J. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol 2005; 58: 594–604. Dalmau J, Tuzun E, Wu HY, et al. Paraneoplastic anti-N-methyl-Daspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61: 25–36. Maarten J Titulaer and all Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study Lancet Neurol 2013; 12: 157–65 Honnorat J, Viaccoz A. New concepts in paraneoplastic neurological syndromes Rev Neurol (Paris). 2011 167(10):729-36.
Troubles de la marche chez un patient traité par cisplatine
Un homme de 56 ans signale des troubles de la marche évoluant depuis environ deux mois. Il se plaint de difficultés occasionnelles à se relever d’une chaise, de crampes des cuisses et d’une constipation d’apparition récente. Il est actuellement traité par cisplatine pour un cancer pulmonaire à petites cellules dont le bilan d’extension est négatif. Il suit par ailleurs un traitement par statines.
L’examen neurologique met en évidence un déficit moteur discret bilatéral des deltoïdes. La marche est normale. Il n’y a pas de trouble sensitif. Les ROT sont faibles.
Un électromyogramme est réalisé.
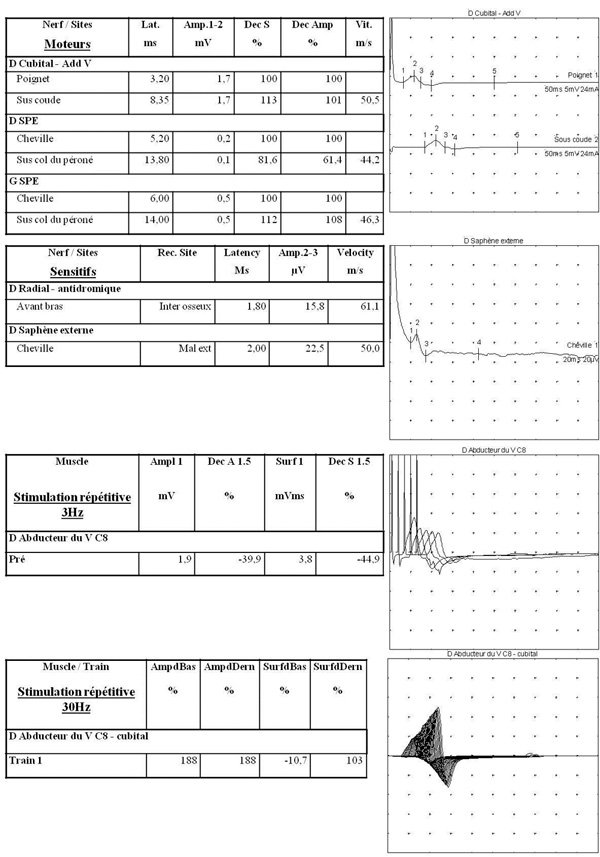
Comment l’interprétez vous ? Quel est votre diagnostic ?
L’EMG met en évidence des réponses motrices de faibles amplitudes tandis que les réponses sensitives sont normales. Un décrément d’amplitude de 40% des potentiels moteurs est observé à basse fréquence (3 Hz) alors qu’à haute fréquence (30 Hz) apparaît une augmentation d’amplitude de 188%.
Il s’agit d’un syndrome de Lambert-Eaton.
Le contexte permet de retenir une origine paranéoplasique.
Le syndrome de Lambert-Eaton correspond à un bloc neuromusculaire pré-synaptique causé par des anticorps anti canaux calciques voltage-dépendants. Il est paranéoplasique dans 60% des cas et concerne alors surtout le cancer pulmonaire à petites cellules.
La triade clinique évocatrice comporte des troubles de la marche, un déficit moteur proximal des membres supérieurs et une dysautonomie (sécheresse muqueuse, constipation). L’atteinte bulbaire est exceptionnelle. L’examen électromyographique met en évidence une augmentation de l’amplitude des potentiels d’action moteurs >100% après effort ou stimulation à haute fréquence.
Le pronostic des formes paranéoplasiques est lié à celui du cancer sous-jacent.
Proposé par : Jean-Paul Bouwyn (Rouen).
Références :
J. Newsom Davis. Lambert-Eaton myasthenic syndrome. Rev Neurol (Paris) 2004 ; 160 :2, 177-180.
Titulaer MJ et al. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011 Dec;10(12):1098-107.
Tremblement intentionnel progressif et asymétrique
Un patient de 68 ans vous consulte pour une dysgraphie. Il n’a pas d’antécédent particulier sur les plans personnel et familial. Son examen objective un tremblement d’intention du membre supérieur droit isolé.
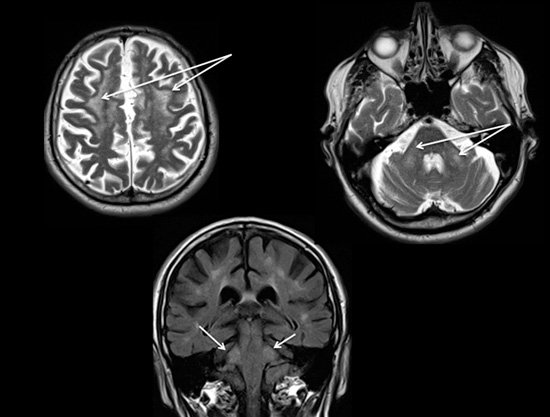
Vous réalisez une IRM cérébrale.
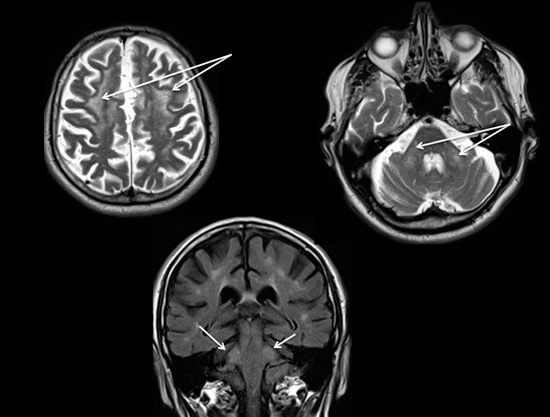
- Quel diagnostic faut il évoquer en priorité ? Comment le recherchez-vous ?
- Votre diagnostic est confirmé, que proposez vous au patient ?
1- L’IRM montre des hypersignaux périventriculaires et des pédoncules cérébelleux moyens symétriques.
Il faut évoquer devant ce tableau clinique et radiologique le diagnostic de FXTAS et demander une recherche moléculaire.
2- Un conseil génétique est indiqué en raison du risque de transmission du syndrome de l’X fragile chez les descendants du patient.
La recherche génétique de prémutation de l’X fragile retrouve un nombre de répétition de triplet CGG estimé à 103 et qui confirme le diagnostic de FXTAS.
Le syndrome FXTAS affecte essentiellement les hommes vers 70 ans. Les signes cliniques majeurs sont l’ataxie cérébelleuse, le tremblement d’intention, le syndrome dyséxécutif, le syndrome parkinsonien et la neuropathie périphérique. La prévalence du syndrome dans cette population est estimée à environ 1 sur 3000. L'IRM cérébrale montre habituellement une atrophie cérébrale globale et des lésions de la substance blanche au niveau des pédoncules cérébelleux moyens. La transmission est liée au chromosome X et est dominante. Le syndrome est dû à une prémutation du gène FMR1 (fragile X mental retardation 1, Xq27.3) qui se caractérise par l'expansion de 55 à 200 triplets CGG. Les individus normaux ont la séquence CGG répétée moins de 40 fois. Les individus atteints du syndrome de l’X fragile ont cette séquence répétée plus de 200 fois et jusqu’à des milliers de fois, entraînant un mauvais fonctionnement de gène responsable de la maladie. Ce diagnostic est la cause la plus fréquente de retard mental et d’autisme.
Les autres leucopathies (dont l’incidence est nettement plus rare) à évoquer devant des hypersignaux symétriques des pédoncules cérébelleux sont le déficit en lamine B1, la leucoencéphalopathie avec atteinte du tronc cérébral et de la moelle avec augmentation de lactate (syndrome LBSL), la leucodystrophie de l’adulte autosomale dominante (ADLD).
Référence :
1- Maureen A. Leehey. Fragile X-associated Tremor/Ataxia Syndrome (FXTAS): Clinical Phenotype, Diagnosis and Treatment. J Investig Med. 2009 December; 57(8): 830–836.
2- Labauge P. Génétique et maladies de la substance blanche, EMC 17-001-A-50.
Un diagnostic facilité par l'imagerie typique
Un jeune homme de 19 ans, d'origine vietnamienne, arrivé depuis peu en France, est hospitalisé pour syndrome confusionnel d'installation progressive depuis quelques semaines. L'examen neurologique montre une rigidité plastique hémicorporelle gauche. Le bilan biologique standart est normal. Une IRM cérébrale est rapidement réalisée.
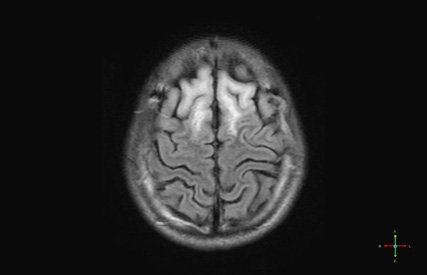
Quel est votre diagnostic ?
Les images IRM sont très évocatrices dans ce contexte de maladie de Wilson :
- Anomalies de signal en T2 et FLAIR simultanées dans les ganglions de la base, les thalami, et le tronc cérébral
- Aspect de « face de Panda géant » sur les coupes axiales T2 et FLAIR: hyperintensité du tronc cérébral, notamment autour de noyaux rouges qui eux restent en hyposignal
- Hypersignal T2 et FLAIR du tectum du mésencéphale
- Les autres anomalies parfois rencontrées sont : un aspect de myélinolyse centropontine, des anomalies de la substance blanche et des hypersignaux corticaux T2 (de pronostic péjoratif)
Bibliographie :
L.K. Prashanth, S. Sinha, DM, A.B. Taly, and M.K. Vasudev, MRI Features Distinguish Wilson's Disease from Other Early Onset Extrapyramidal Disorders? An Analysis of 100 Cases Mov Disord 2010, pp. 672–678 Sinha S, Taly AB, Prashanth LK, Ravishankar S, Arunodaya GR, Vasudev MK Sequential MRI changes in Wilson's disease with de-coppering therapy: a study of 50 patients The British Journal of Radiology, 80 (2007), 744–749
Syndrome cérébelleux subaigu
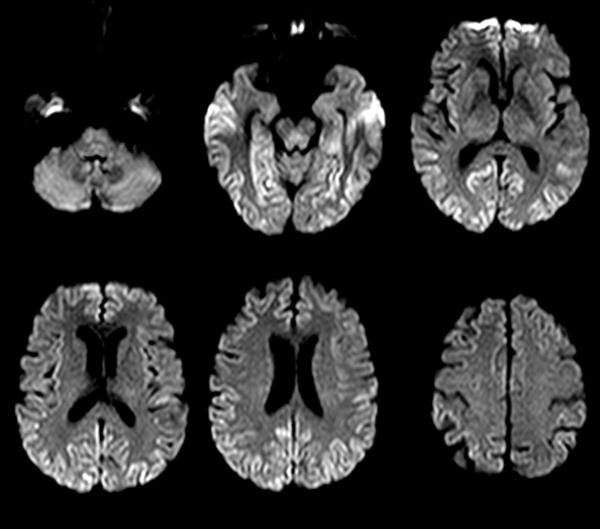
Une femme de 58 ans, sans antécédant, employée chez Air France, est hospitalisée pour des troubles de la marche et de l'équilibre apparus de manière progressive depuis 3 mois. L'interrogatoire révèle également des troubles visuels sous la forme d'une diplopie intermittente. L'examen neurologique montre un syndrome cérebelleux axial, une discrète dysarthrie, et une limitation des saccades oculaires. Il n'y a pas d'atteinte des fonctions cognitives. Une IRM cérébrale est rapidement réalisée.
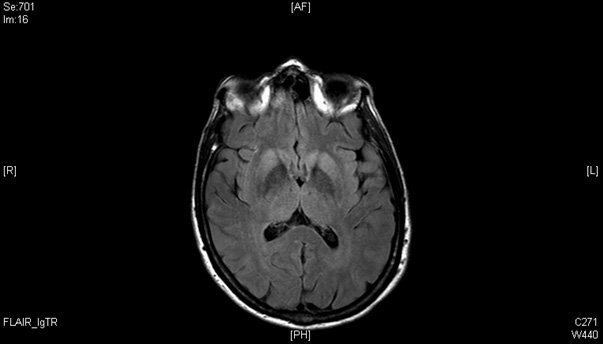
Quel diagnostic évoquez-vous ?
L'histoire clinique couplée à l'hypersignal en diffusion et en FLAIR des noyaux gris permet d'évoquer le diagnostic de maladie de Creutzfeldt Jakob.
Les lésions IRM évocatrices de la maladie de Creutzfeldt Jakob sont :
- sur les séquences en diffusion : un hypersignal cortical, du striatum, ou plus rarement du thalamus
- sur les séquences en FLAIR : un hypersignal des mêmes territoires (striatum, cortex, thalamus), mais qui peut être plus tardif
Bibliographie :
Vitali P, Maccagnano E, Caverzasi E, Henry RG, Haman A, Torres-Chae C, Johnson DY, Miller BL, Geschwind MD. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011;76(20):1711-9 Macfarlane RG, Wroe SJ, Collinge J, Yousry TA, Jäger HR. Neuroimaging findings in human prion disease. J Neurol Neurosurg Psychiatry. 2007;78(7):664-70
Une lésion cérébrale isolée
Un homme de 22 ans sans antécédent, a constaté au réveil le 12 juin 2010 une lourdeur du membre supérieur gauche, complétée 3 jours plus tard d'une hémiparésie gauche motrice pure; il ne pouvait pas se lever seul. L'étude du LCR retrouvait : protéines 0.44 g/L, 6 lymphocytes /mm3, recherche de bandes oligoclonales positive.
L'IRM cérébrale retrouvait les anomalies suivantes :
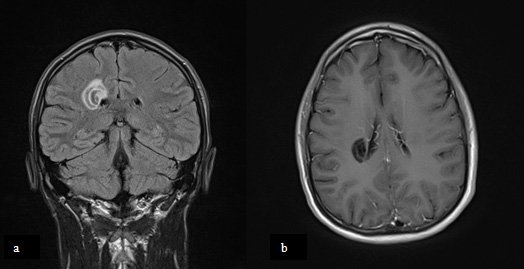
Quel est le diagnostic?
Il s'agit d'une sclérose concentrique de Balo, forme rare de SEP. Cette forme particulière de la maladie souvent d'installation aiguë peut avoir une évolution monophasique et être de mauvais pronostic en cas de lésions diffuses mais peut évoluer de façon chronique avec rémissions des symptômes. Elle touche surtout l'adulte jeune avec une prédominance masculine et est surtout décrite en Asie.
Radiologiquement, les lésions ont un aspect concentrique et sont composées de zones hypointenses T1 et hyperintenses T2 correspondant à une nécrose cavitaire avec démyélinisation de la substance blanche et d'anneaux isointenses en T1 et T2 correspondant à de la myéline normale. Il peut exister un réhaussement des lésions plus ou moins partiel après injection de Gadolinium.
Elles sont typiquement localisées au niveau de la substance blanche frontale et pariétale. Le tronc cérébral, le cervelet et le cordon médullaire peuvent aussi être touchés.
Bibliographie :
De Seze J. Formes frontieres de la sclérose en plaques. Rev Neurol (Paris). 2006 Jan;162(1):137-43. - Kreft KL, Mellema SJ, Hintzen RQ. Spinal cord involvement in Balo's concentric sclerosis. J Neurol Sci. 2009 Apr 15;279(1-2):114-7.
Coma aigu dans un contexte d'alcoolisme
Un Homme de 53 ans, alcoolique chronique, avait été hospitalisé une première fois pour confusion mentale et troubles de l'équilibre, aigus, régressifs en 8 jours, sans explication trouvée. Il est réhospitalisé 4 mois plus tard en raison d'un coma aigu : cet homme, désocialisé, a été retrouvé à son domicile.
Voici son IRM cérébral :
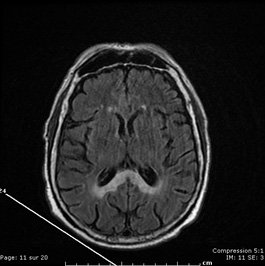
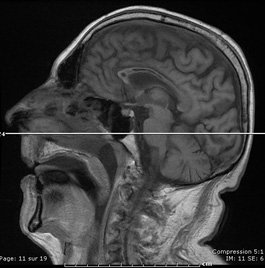
Quel est diagnostic ?
Il s'agit d'une maladie de Marchiafava-Bignami associée à un début de myélinolyse centro pontique.
La maladie de Marchiafava Bignani est une complication rare de l'alcoolisme, caractérisée par une démyélinisation et une nécrose du corps calleux. Les signes cliniques sont variables : confusion, troubles de la conscience, troubles cognitifs, tétraparésie, astasie abasie, signes de dysconnexion calleuse…
Son pronostic est en général mauvais, souvent conditionné par l'importance de l'atteinte du corps calleux L'IRM cérébrale montre en T2 et FLAIR des hypersignaux plus ou moins diffus du corps calleux.
Bibliographie :
Uchino A, Takase Y, Nomiyama K, Egashira R, Kudo Acquired lesions of the corpus callosum: MR imaging.S Eur Radiol. 2006 Apr;16(4):905-14..
Heinrich A, Runge U, Khaw AV. Clinicoradiologic subtypes of Marchiafava-Bignami disease. J Neurol. 2004 Sep;251(9):1050-9.
Ornithologie et neurologie
Un homme de 70 ans, sans autre antécédent qu’une cataracte bilatérale, vint en consultation pour des troubles de la marche et une gêne visuelle mal définie.
Les troubles de la marche avaient débuté 5 ans auparavant par des festinations et il existait alors un tremblement de repos des membres supérieurs. Les troubles avaient fait évoquer une maladie de Parkinson et avaient bien répondu à la dopathérapie (lévodopa 3x 150 mg/j). Depuis deux ans il présentait un ralentissement de la marche, qui se faisait à petits pas, avec une raideur axiale et une instabilité posturale. L’attitude se faisait en antéflexion. Le visage était amimique, il n’y avait pas de tremblement, de syndrome akinéto-hypertonique segmentaire, de signe de dysautonomie, ni de signes pyramidaux. Les mouvements de verticalité du regard étaient diminués vers le haut.
Le patient obtenait un score de 26/30 au MMSE de Folstein. Les troubles de la mémoire étaient dus à des troubles du rappel. Il existait de nettes difficultés dans les tâches d’évocation catégorielle, une tendance aux persévérations parasitant la programmation motrice, une approbation en écho. Les fonctions visuo-perceptives et visuo-praxiques étaient bien préservées.
Une IRM cérébrale avait été réalisée avant la consultation neurologique et n’avait pas évoqué au radiologue de diagnostic particulier (figures 1 et 2).
Partagez-vous cette analyse ? Que faut-il y voir ?
Quel diagnostic retenez-vous ?
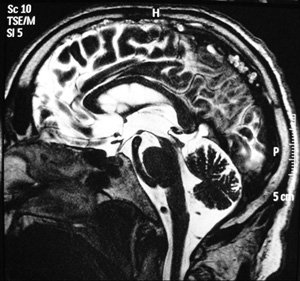
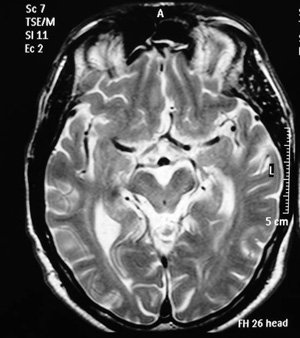
Figure 2 - IRM cérébrale. Coupe axiale. Séquence pondérée en T2.
1. Le signe du colibri et le signe de l’ipomée (morning glory sign). Voir figure ci-dessous.
Le « signe du colibri », désigné par d’autres auteurs « signe du pingouin », s’observe en IRM morphologique sur des coupes sagittales médianes [1]. Il est lié à une perte du volume du mésencéphale épargnant relativement le pont, avec une perte de la convexité du tegmentum mésencéphalique, qui devient rectiligne voire concave vers le haut. Le mésencéphale rappelle alors la tête d’un colibri ou d’un pingouin, alors que le volume préservé du pont en figure le ventre.
Le « signe de l’ipomée » s’observe en IRM sur des coupes axiales du mésencéphale. La concavité des bords latéraux du tegmentum mésencéphalique et l’élargissement du sillon interpédonculaire donnent une image rappelant la forme biconcave de l’ipomée, variété de fleur dont la plus commune sous nos latitudes est le liseron. Certains auteurs américains parlent aussi de « signe de Mickey Mouse », par analogie avec les oreilles de Mickey.
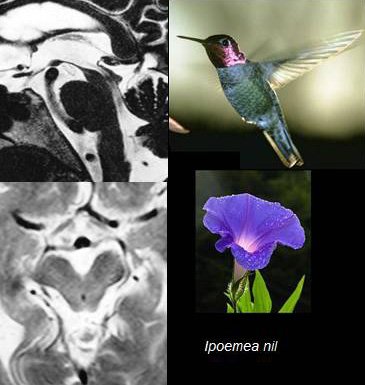
(image en haut à gauche).
Sur la coupe axiale du tronc cérébral, la concavité des bords latéraux du tegmentum mésencéphalique et l’élargissement du sillon interpédonculaire suggèrent la forme d’une ipomée (morning glory sign, signe de l’ipomée) ou des oreilles de Mickey Mouse (image en bas à gauche).
2. Ces deux signes sont très évocateurs d’une paralysie supranucléaire progressive (PSP). Le signe du colibri aurait une sensibilité de 68% et une spécificité de 88,6%. Le signe de l’ipomée aurait une sensibilité de 68% et une spécificité de 77,7% [2].
On sait depuis quelques années que certaines PSP ne se conforment pas au tableau classique popularisé par Steele, Richardson et Olszewski, et présentent un syndrome parkinsonien initial avec tremblement de repos voire une réponse positive à la dopa [3]. C’était le cas chez ce patient, dont le tableau n’est devenu évocateur d’une PSP qu’après quelques années d’évolution.
Auteurs :
François Sellal, Christophe ZaenkerDépartement de Neurologie, Hôpitaux Civils de Colmar, 68024 Colmar Cédex, France.
Références :
[1]. OBA H, YAGISHITA A, TERADA H, BARKOVITCH AJ, KUTOMI K, YAMAUCHI T. et al. New and reliable MRI diagnosis for progressive supranuclear palsy. Neurology 2005; 64: 2050-5
[2]. RIGHINI A, ANTONINI A, DE NOTARIS R, BIANCHINI E, MEUCCI N, SACILOTTO G et al. MR Imaging of the Superior Profile of theMidbrain: Differential Diagnosis between Progressive Supranuclear Palsy and Parkinson Disease. AJNR 2004; 25: 927-32.
[3]. WILLIAMS DR, LEES AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol 2009; 8: 270–9.
Le déficit est de rigueur
Un homme de 76 ans est admis aux urgences devant la succession de plusieurs épisodes déficitaires sensitifs touchant son membre supérieur gauche, survenus au cours des derniers jours. Il présente dans ses antécédents une hypertension artérielle ancienne et traitée, un adénocarcinome prostatique traité par hormonothérapie et radiothérapie en rémission et une hémorragie sous arachnoïdienne post traumatique frontale droite survenue 3 ans auparavant.
A sa prise en charge aux urgences, il présente une hypoesthésie brachiofaciale gauche ainsi qu’une dysarthrie. Ces symptômes régressent rapidement et sont identiques à ceux qu’il ressentait depuis une semaine. Le scanner cérébral réalisé en urgence met en évidence une leucoaraïose périventriculaire. L’exploration des troncs supra-aortiques note une plaque athéromateuse carotidienne gauche. L’éléctroencéphalogramme est normal. L’IRM encéphalique vous est fournie (figure 1).
Quel est votre diagnostic ?
Olivier Casez
Pôle de Psychiatrie et de Neurologie, CHU GRENOBLE, 30843 Grenoble cedex 9 - OCasez@chu-grenoble.fr
Diagnostic : crises épileptiques partielles symptomatiques d’une sidérose méningée séquellaire de son HSA. En effet, on peut constater sur l’IRM pondérée en T2* un hypersignal au sein des sillons central, précentral et frontal antérieurs droits. Il n’y a pas d’anomalie sur les séquences FLAIR, écartant un saignement récent. Les épisodes neurologiques de nature déficitaire peuvent également se rencontrer au cours des crises épileptiques. Le traitement antiépileptique a permis de faire disparaître l’ensemble des symptômes.
Références :
Lüders HO, Dinner DS, Morris HH, et al (1995). Cortical electrical stimulation in humans. The negative motor areas. Adv Neurol., 67: 115-29.
Lüders H, Acharya J, Baumgartner C, et al (1998). Semiological seizure classification. Epilepsia, 39(9): 1006-13.
Noguchi K, Ogawa T, Seto H, et al (1997). Subacute and chronic subarachnoid hemorrhage: diagnosis with fluid-attenuated inversion-recovery MR imaging. Radiology, 203(1): 257-62.
Une maladie sous le feu des projecteurs
Un homme de 65 ans sans antécédent médical personnel ni familial consulte pour une apathie marquée ainsi que des difficultés dans les actes de la vie quotidienne (habillage, lavage) qui évoluent depuis environ deux mois et qui viennent compliquer un état dépressif résistant à une prise en charge médicamenteuse et psychothérapique, évoluant depuis 4 mois.
L’examen neurologique ne relève pas de déficit focalisé sensitif ni moteur, pas de syndrome parkinsonien. L’évaluation neuropsychologique réalisée met en évidence une aphasie antérieure, un trouble du discours de type frontal ainsi qu’une apathie majeure sans tristesse de l’humeur ainsi que des troubles praxiques. Il n’y a pas d’hallucination. On ne note pas de fluctuation de son état clinique. Une IRM est alors réalisée et vous est présentée (figure 1).
Quel diagnostic vous suggère ces résultats d’imagerie ? Comment affinez-vous votre diagnostic et comment le confirmer ?
Diagnostic : maladie de Creutzfeldt-Jakob. L’IRM encéphalique de diffusion met en évidence un hypersignal de l’ensemble du ruban cortical très évocateur dans ce contexte d’une maladie de Creutzfeldt Jakob. L’EEG peut mettre en évidence un tracé pseudopériodique à 1 Hz diffus. La recherche de la protéine 14.3.3, marqueur d’une lyse neuronale accélérée, est classiquement positive. La certitude diagnostique est apportée par l’examen anatomopathologique de l’encéphale et la recherche de la protéine prion par western blot au sein de ce tissu.
Cas proposé par : Olivier Casez
Pôle de Psychiatrie et de Neurologie, CHU GRENOBLE, 30843 Grenoble cedex 9 - OCasez@chu-grenoble.fr
Références :
Kallenberg K, Schulz-Schaeffer WJ, Jastrow U, et al (2006). Creutzfeldt-Jakob disease: comparative analysis of MR imaging sequences. AJNR Am J Neuroradiol, 27: 1459-1462.
S. Nougaret, H. Brunel, G. Bourbotte, et al (2007). Diffusion-weighted MRI in sporadic Creutzfeldt-Jakob disease. J Neuroradiol, 34(4): 260-6.
Un indice s'il vous plaît !
Une femme de 58 ans, au principal antécédent de polykystose rénale, est prise en charge en urgence pour une hémorragie sous arachnoïdienne sur anévrysme de la communicante antérieure compliquée initialement d’une hydrocéphalie. L’embolisation initiale se complique d’un hématome interhémisphérique se résorbant au cours des quinze jours suivants. Elle quitte le service de réanimation au bout de trois semaines pour la neurologie avec un syndrome « confusionnel ».
À sa prise en charge en neurologie, on note une désorientation temporospatiale, des troubles mnésiques et un syndrome fronto-dysexécutif. Il n’y a pas de trouble de la vigilance. Son examen neuropsychologique met en évidence les éléments suivants : un syndrome amnésique avec un trouble de la mémoire antérograde très amélioré par l’indiçage (sur matériel verbal et visuospatial), de nombreuses fabulations. Il s’y associe un syndrome frontal dysexécutif dominé par des persévérations et des difficultés d’évocation.
L’imagerie encéphalique (IRM, TDM cérébrale de perfusion) réalisé à trois semaines de l’HSA, met en évidence une évolution favorable de l’hématome inter-hémisphérique, l’absence de spasme artériel ou de lésion cérébrale survenue dans l’intervalle.
Elle n’a jamais consommé d’alcool ni d’autres toxiques. Son bilan biologique métabolique est sans particularité.
Quel diagnostic syndromique neuropsychologique avez-vous fait ?
Diagnostic : syndrome amnésique par lésion du « basal forebrain » (cerveau basal antérieur). Cette patiente présente un syndrome amnésique par lésion du basal forebrain (région basale sous frontale, cf. figure 1 : région comprenant le noyau basal de Meynert, les noyaux du septum, les bandelettes diagonales de Broca). Il s’agit d’une entité syndromique associant une amnésie antérograde sévère très sensible à l’indiçage, une amnésie rétrograde variable et des fabulations importantes. Il peut s’y associer un syndrome fronto-dysexécutif. Les premiers patients décrits avaient présenté ce type de troubles suite d’hémorragie sous arachnoïdienne sur rupture d’anévrysme de la communicante antérieure. L’évolution est généralement de pronostic relativement favorable avec critique puis disparition des fabulations et plus progressivement, amélioration du syndrome amnésique et du syndrome frontal.
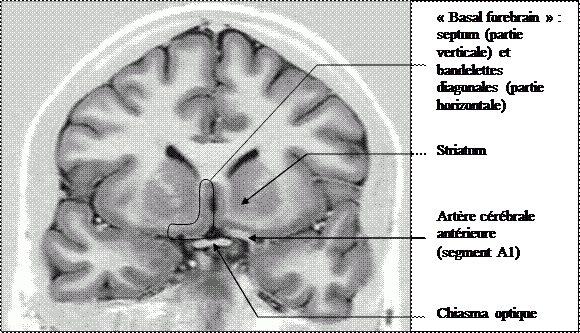
Cas proposé par : Olivier Casez
Pôle de Psychiatrie et de Neurologie, CHU GRENOBLE, 30843 Grenoble cedex 9 - OCasez@chu-grenoble.fr
Références :
Damasio AR, Graff-Radford NR, Eslinger PJ, et al (1985). Arch Neurol., 42(3): 263-71.
Damasio AR, Eslinger PJ, Damasio H, et al (1985). Multimodal amnesic syndrome following bilateral temporal and basal forebrain damage. Arch Neurol., 42(3): 252-9.
Céphalée brutale et récidivante
Un homme de 49 ans a présenté en 2008, au volant de sa voiture, une céphalée brutale en hémicrânie, régressive sous antalgiques. Le lendemain, la céphalée récidive à l’othostatisme et n’est plus calmée par les antalgiques. Elle régresse par contre en position couchée.
L’examen clinique n’objective pas d’anomalie hormis cette céphalée positionnelle.
L’interrogatoire retrouve la notion d’une fracture de côte basi-thoracique gauche suite à un choc 2 ans auparavant.
Un TDM cérébral sans et avec injection de produit de contraste est considéré comme normal. L’évolution 3 jours plus tard est marquée par l’installation d’une diplopie binoculaire par paralysie du VI bilatérale.
La prise de contraste méningée évoque le diagnostic d’hypotension spontanée du liquide céphalo-rachidien (LCR) . Elle est due à une hypovolémie du LCR. Il existe souvent dans les jours précédents, un contexte de traumatisme rachidien mineur, d’activité sportive ou d’efforts inhabituels ou parfois une notion de maladie du tissu élastique qui va se compliquer d’une brèche durale.
Les céphalées sont sévères, installées plus ou moins progressivement, parfois brutalement ou à l’occasion d’un effort. Elles sont pulsatiles occipitales irradiant vers les épaules ou le rachis cervical, à l’orthostatisme et disparaissent en décubitus après quelques minutes. Il s’y associe des nausées, vomissements, acouphènes ou diplopie par atteinte du VI.
Si la céphalée devient permanente, deux complications sont à rechercher : la thrombophlébite cérébrale et l’hématome sous-dural compressif.
La confirmation diagnostique est basée sur l’IRM cérébrale notamment les séquences T1 sans et avec injection de gadolinium qui visualisent une prise de contraste diffuse des pachyméninges supra et infra-tentorielles s’étendant parfois jusqu’à la moelle cervicale. Ces anomalies peuvent manquer dans 1/3 des cas. L’IRM peut mettre en évidence des hématomes ou des hydromes sous-duraux, un effacement des citernes prépontiques et chiasmatiques et de petits ventricules. La recherche de la brèche n’a que peut d’intérêt en pratique.
Le traitement repose sur les mesures symptomatiques ainsi que la réalisation parfois à répéter d’un blood-patch épidural lombaire.
Pour en savoir plus :
Mokri B(1999) Mayo Clin Proc. 1999 Nov;74(11):1113-23. Bousser MG (2005) Rev Neurol (Paris) ; 6-7: 700-702
Chan EK (2011) J Child Neurol ; 26(6) : 761-766
Piège diagnostique
Un homme de 62 ans aux antécédents de surcharge pondérale, de tabagisme sevré, d’HTA se plaignait de lombo-sciatalgies bilatérales résistantes au traitement antalgique. Trois semaines plus tard, il est hospitalisé devant l’installation d’une rétention aiguë d’urine et de troubles de la marche dans un contexte sub-fébrile.
L’examen clinique objectivait un déficit sensitivo-moteur proximal et distal des membres inférieurs asymétrique prédominant à gauche, des réflexes ostéo-tendineux abolis aux membres inférieurs, des réflexes cutanés plantaires indifférents, une hypoesthésie en selle et un globe vésical nécessitant un sondage urinaire.
Une première IRM lombaire réalisée sans injection de gadolinium à la recherche d’une spondylodiscite ne retrouvait que des discopathies L4-L5, L5-S1 non pathogènes.
Une PL retrouvait une hyperprotéinorachie (2g/l) et une réaction lymphocytaire (66 lymphocytes/mm3).
Une nouvelle IRM était réalisée quelques jours plus tard devant l’aggravation de l’état du patient malgré une antibiothérapie débutée après la PL.
Qu'en pensez-vous ?
L’aspect IRM est évocateur d’une fistule artério-veineuse durale de la moelle épinière.
Il s’agit de la malformation vasculaire de la moelle épinière la plus commune.
Les symptômes associent dans la forme complète un déficit sensitivo-moteur des membres inférieurs et des troubles sphinctériens. Une douleur dorsale peut être présente. Le tableau peut parfois mimer une atteinte du second motoneurone. Les hommes de plus de 60 ans sont les plus concernés. Les symptômes peuvent évoluer souvent sur plusieurs mois ou années mais la présentation peut être aussi subaiguë.
La fistule correspond à une connexion anormale entre une branche d’une artère et une veine radiculaires durales, le plus souvent au niveau thoracique bas ou lombaire. Une augmentation de la pression veineuse puis une congestion veineuse intra-médullaire résultent de ce shunt. Cette congestion se traduit pas une myélopathie voire un infarctus médullaire qui explique la symptomatologie.
L'IRM médullaire met en évidence une augmentation du calibre médullaire avec un hypersignal T2 intra-médullaire s'étendant sur plusieurs segments vertébraux traduisant l'œdème (fig. 1).On peut noter une dilatation des vaisseaux péri-médullaires plutôt sur la face dorsale de la moelle qui peuvent être déjà visibles en hypersignal T2 (effet d’absence de signal ou « flow void » ) ou au moins réhaussés après injection de gadolinium (fig.2).
L'artériographie peut confirmer le diagnostic si besoin en mettant en évidence les vaisseaux dilatés tortueux et le shunt.
Le traitement repose sur l'embolisation par voie endovasculaire de la veine durale incriminée ou une prise en charge chirurgicale. La morbidité semble équivalente avec les 2 procédures, La durée d'hospitalisation est plus courte après traitement endovasculaire mais au prix d'un risque de récurrence plus important.
Pour en savoir plus :
Krings T. et al. Spinal dural arteriovenous fistulas. Am J Neuroradiol. 2009 ;30 :639-648
Narvid J. Spinal dural arteriovenous fistulae : Clinical Features and Long-term results. Neurosurgery.2008;62:159-167
Cho K.T. et al. Treatment of spinal cord perimedullary arteriovenous fistula : embolization versus surgery. Neurosurgery.2005;56:232-239
Gilberston J. et al. Spinal dural arteriovenous fistulas : MR and myelographic findings, AJNR. 1995;16:2049-2057
Crise convulsive chez une patiente malgache
Une femme de 25 ans, d’origine malgache, s’est présenté aux Urgences pour un syndrome fébrile initialement attribué à une infection urinaire. Un traitement par ceftriaxone a été proposé. Le lendemain, elle est hospitalisée en raison de deux crises convulsives inaugurales. L’examen neurologique est normal, mais la patiente signale des myalgies diffuses.
Un scanner cérébral, puis une IRM cérébrale sont rapidement réalisées.
Quelle est votre hypothèse diagnostique ?
On suspecte une neurocysticercose devant l’association de crises convulsives, d’une lésion corticale frontale gauche avec œdème péri-lesionnel chez une patiente d’origine malgache.
La neurocysticercose est principalement retrouvée dans les pays dont les conditions d’hygiène sanitaire sont insuffisantes.
Le parasite incriminé est un ver, le Taenia solium, dont les œufs sont absorbés accidentellement notamment en mangeant de la viande de porc contaminée, insuffisamment cuite.
Les larves issue de ces œufs migrent dans l’organisme avec une prédilection pour les organes richement vascularisés notamment le SNC.
Les symptômes cliniques vont dépendre de la localisation des kystes (parenchyme, ventricules, méninges, moelle épinière). Les localisations extra-parenchymateuses sont surtout retrouvées chez l’adulte.
Les plus courantes manifestations sont les crises convulsives (partielles± généralisées) dans 70 à 90% des cas. Les autres modes de présentation associent des céphalées, déficits focaux , hypertension intracrânienne, syndrome méningé, atteinte des nerfs crâniens, ataxie, troubles visuels voire troubles du comportement.
Le diagnostic repose sur en premier lieu le TDM cérébral qui met en évidence une ou plusieurs lésions de petite taille (< 20 mm) hypodenses avec un réhaussement en couronne après injection et un œdème péri-lésionnel (Fig 1). Il existe souvent une calcification au sein du kyste punctiforme ou de très petite taille. En IRM, la lésion apparaît en iso- ou relatif hypersignal et est parfois mieux visualisée avec l’œdème périlésionnel en hypersignal en FLAIR. Le même réhaussement en couronne est noté après injection (Fig 2). Le diagnostic différentiel avec un tuberculome dans les régions d’endémie peut se poser mais les lésions sont généralement plus volumineuses, de contours plus irréguliers et l’œdème plus sévère.
Un diagnostic sérologique est disponible mais n’a pas une sensibilité de 100%
La prise en charge thérapeutique repose sur le traitement anti-parasitaire (albendazole ou praziquantel) qui réduisent le nombre et la taille des lésions auquel peut être associé une corticothérapie et un traitement symptomatique.
En cas de lésion unique, le pronostic est généralement bon avec une disparition des images en 6 mois dans 60% des cas. En cas de lésions multiples ou de lésions calcifiées, les crises d’épilepsie peuvent récidiver fréquemment malgré le traitement. Les localisations extra-parenchymateuses ont un pronostic plus réservé.
Pour en savoir plus :
Carabin et al. Clinical manifestations associated with neurocysticercosis : a sytematic review. PloS Negl Trop Dis 2011 ; 5 (5):e1152
Singhi. Neurocysticercosis. Ther Adv Neurol Disord. 2011; 4(2):67-81
Modifications de signal intra-médullaire
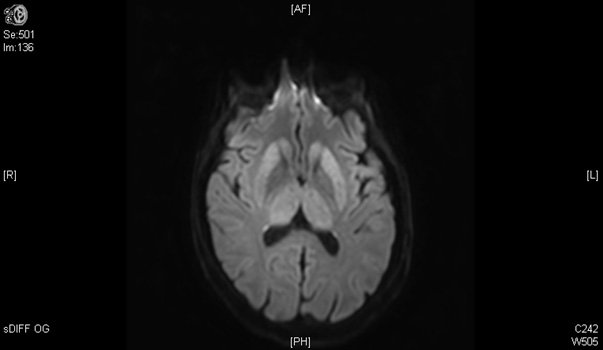
Une femme de 50 ans, sans antécédent, fut hospitalisée pour le bilan de troubles sensitifs des 4 membres évoluant depuis 1 mois. Elle décrivait des troubles de l’équilibre et une sensation d’engourdissement et de peau cartonnée des deux pieds s’étant étendue progressivement au tronc puis au deux membres supérieurs.
L’examen clinique montrait une démarche ataxique, des troubles de la kinesthésie et de la graphesthésie ainsi qu’une hypopallesthésie aux 4 membres ; on notait une faiblesse des 4 membres, des réflexes ostéo-tendineux conservés et un réflexe cutané plantaire indifférent à gauche. Il n’y avait pas de déficit thermo-algique, pas de trouble sphinctérien ni de douleurs rachidiennes.
L’IRM médullaire réalisée est la suivante :
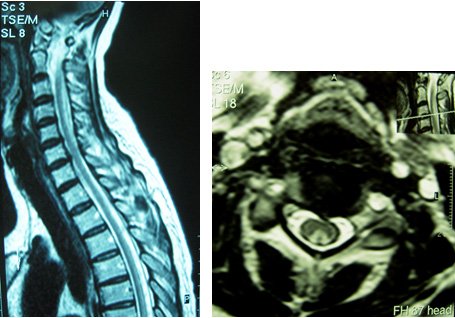
Quelle est votre diagnostique ?
Hypersignal intramédullaire, en séquence T2, des cordons postérieurs de la moelle, étendu de C2 à C5 visible en coupes sagittale et transversale.
Cette atteinte cordonnale postérieure clinique et radiologique est très évocatrice d’une carence en vitamine B12. Associée à un atteinte des cordons latéraux, elle réalise le tableau typique de sclérose combinée de la moelle qui constitue une cause curable classique mais rare d’atteinte médullaire progressive de l’adulte.
La carence en cuivre, beaucoup plus rare, peut se traduire par un tableau clinique et radiologique similaire.
Pour en savoir plus :
Mankad K, Kullmann DM, Davagnanam I . Neurological manifestation of vitamin B12 deficiency. Am J Med. 2010 Jun;123(6):e1-2.
Un LCR jaune comme de l'urine
C’est un homme de 59 ans qui présente une ataxie cérébelleuse progressive apparue il y a 5 à 6 ans. Il a également une hypoacousie bilatérale prédominant à droite, pour laquelle il a été appareillé.
Dans ses antécédents, on retient un traumatisme crânien en 1986 grave responsable d'un coma, avec un arrachement du plexus brachial à droite pour lequel il a été traité avec mise en place d’agrafes.
L’examen neurologique retrouve une ataxie cérébelleuse avec un élargissement du polygone de sustentation et une hypermétrie des deux membres inférieurs. Le patient marche habituellement avec une canne.
L’IRM encéphalique est réalisée. Deux images en séquence écho de gradient pondérée T2 vous sont données.
Le LCR retrouvait un liquide xanthochromatique (« jaune comme de l’urine » selon le patient) avec des sidérophages.
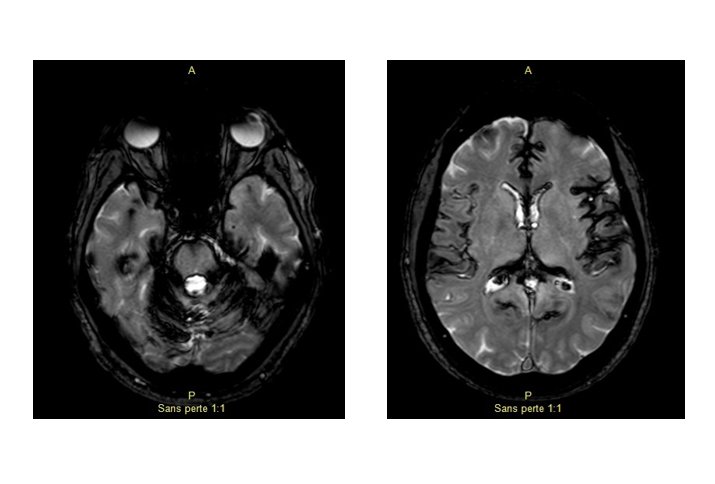
Quel est votre diagnostic?
Le diagnostic est celui d’hémosidérose marginale également appelée sidérose superficielle du système nerveux central. Il s’agit d’un syndrome clinico-radiologique rare caractérisé par des dépôts d’hémosidérine dans les leptoméninges et dans les régions sous-piales.
La présentation clinique classique est une triade associant une ataxie cérébelleuse, une surdité de perception bilatérale et une myélopathie avec syndrome pyramidal fréquemment limitée à une hyper-réflexie. Un déclin cognitif peut être retrouvé.
L’hémosidérose marginale peut être secondaire à une cause de saignement occulte et répété dans les espaces sous arachnoïdiens dont l’origine peut être:
- Une tumeur cérébrale ou rachidienne (21% des cas)
- Un trauma crânien ou vertébral (13 % des cas)
- Un traumatisme du plexus brachial (6% des cas)
- Une malformation vasculaire (9% des cas)
- Un antécédent de neurochirurgie (7% des cas)
- Une angiopathie amyloïde (3% des cas)
Dans 35 à 50% des cas, il n’y pas de cause retrouvée.
A l’IRM, l’hémosidérine est mise en évidence sous la forme d’hyposignaux en séquence écho de gradient pondérée T2
Le scanner cérébral est peu sensible. Il peut montrer un liseré spontanément hyperdense autour du tronc cérébral qui est évocateur du diagnostic
Le liquide cérébro-spinal est anormal dans 75 %, il peut être xanthochromatique ou hémorragique et son analyse peut retrouver une hyperprotéinorachie, une élévation des taux de fer et de ferritine ainsi que la présence de sidérophages
L'évolution se fait habituellement vers une lente dégradation clinique en l'absence de traitement étiologique d’où l’intérêt de la recherche active d’une cause de saignement.
Dans le cas de notre patient nous complétons l’exploration l’étiologique par une IRM médullaire qui n’a pas retrouvé de cause de saignement accessible à un traitement.
L’hémosidérose marginale est potentiellement sous-diagnostiquée. La réalisation des IRM permettra vraisemblablement de retrouver davantage de cas similaires. Il faudra garder en mémoire la présentation clinique et les images radiologiques
Références:
-Kondziella D1, Lindelof M, Haziri D, Larsen VA, Kruse A. Diagnostic and therapeutic challenges in superficial CNS siderosis. Dan Med J. 2015;62(5). pii: A5079.
-Levy M et al., Superficial siderosis: a case report and review of the literature, Nature Clinical Practice Neurology, (2007) 3, 54-58
-Lannareix V1, Basseka H, Catalaa I, Sevely A, Marson F, Manelfe. Superficial siderosis of the central nervous system: a case report. CJ Radiol. 2002:1850-2.
Un signal recueilli au-dessus de l'oreille
Quel signal est recueilli à partir de ce casque support pour un capteur fixé au niveau de l’écaille temporale gauche, au-dessus de l’arcade zygomatique ?
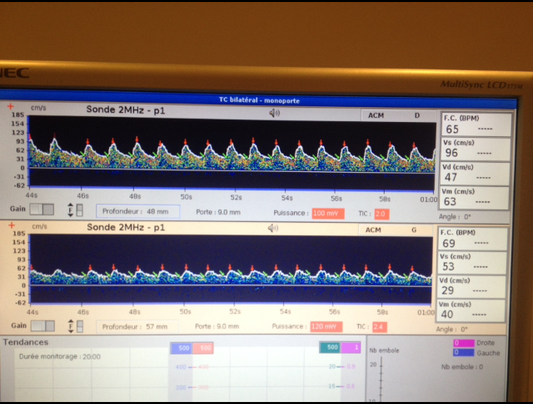
Cérébrales par Doppler trans-crânien
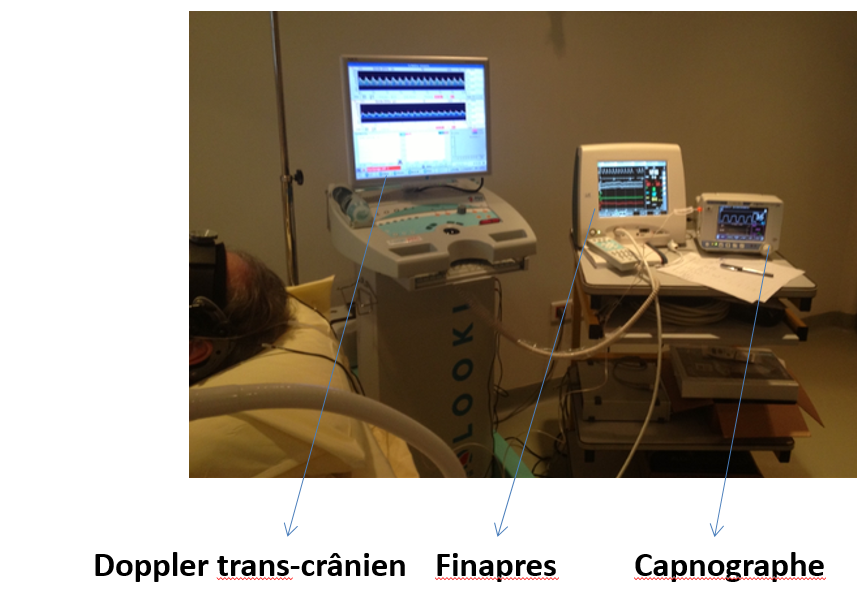
Il s’agit du signal « vitesse circulatoire cérébrale » au niveau de l’artère cérébrale moyenne gauche recueilli de manière continue par Doppler trans-crânien en mode monitoring. Dans ce cas il s’agit une évaluation de la réactivité cérébro-vasculaire au Diamox mesurant la réserve vasomotrice cérébrale en aval d’une sténose de la carotide. L’évaluation intègre le monitoring de la pression artérielle (Finapres) et du CO2 de fin d’expiration (capnographe).
Un indice : les acides gras
- Homme de 42 ans, porteur d’une maladie d’Addison
- Troubles de la marche depuis 1 an
- ATCD fam
- iliaux : 2 demi-frères avec troubles de la marche (1 avec épilepsie)
- Examen clinique :
- Paraparésie spastique
- Hypoesthésie distale symétrique MI
- Syndrome cérébelleux
- Poursuite saccadique
- IRM cérébrale (FLAIR, et T1 Gd)
Quel est votre diagnostic ?
L’adrénoleucodystrophie (ALD)
C’est une maladie génétique récessive liée à l’X qui atteint principalement la substance blanche, les corticosurrénales et les testicules.
Elle est responsable d’un défaut du catabolisme dans le peroxysome des acides gras à très longue chaîne. La maladie comporte une grande variabilité phénotypique.
La forme cérébrale de l’enfant est la plus fréquente et représente environ la moitié des cas ; elle survient après un développement psychomoteur normal mais entraîne une détérioration cérébrale rapidement progressive conduisant à un état végétatif en deux ans.
La seconde forme la plus fréquente est l’adrénomyéloneuropthie de l’adulte (25 %), secondaire à la démyélinisation des cordons postérieurs et antérieurs de la moelle tandis que la substance blanche cérébrale est respectée. Les symptômes neurologiques apparaissent en moyenne à l’âge de 30 ans sous la forme d’une paraparésie spastique et d’une neuropathie périphérique modérée.
Les autres formes, plus rares, sont l’atteinte cérébrale de l’adolescent (5 %), de l’adulte (3 %)
et une insuffisance surrénalienne isolée (10 %).
De plus, certains sujets sont porteurs de l’anomalie génétique mais restent asymptomatiques (8 %).
On classera à part les sujets hétérozygotes symptomatiques : quinze à vingt pour cent des femmes hétérozygotes pour l’ALD peuvent développer une paraparésie spastique progressive, une neuropathie périphérique, des troubles sphinctériens se manifestant en moyenne à 43 ans (+/– 11 ans).
Référence : Desloques et al. 2006
Une cause de multinévrite
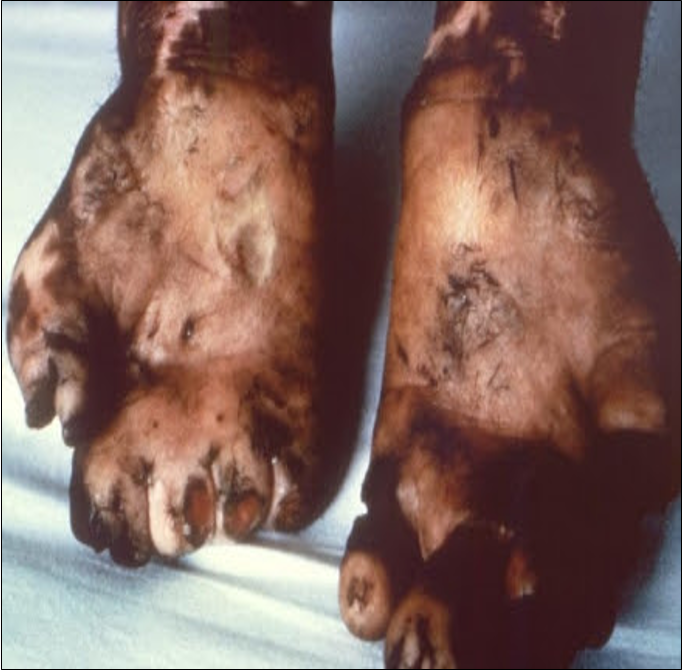
- Patient de 38 ans SDF, africain
- Pas de suivi médical
- Hospitalisé suite à un traumatisme sur la voie publique.
- Examen neurologique : insensibilité des 4 extrémités - mutilations
- Scanner cérébral et IRM cervicale normaux
Quelle est votre suspicion diagnostique ?
Quel est l’examen qui permettrait d’avoir un diagnostic de certitude ?
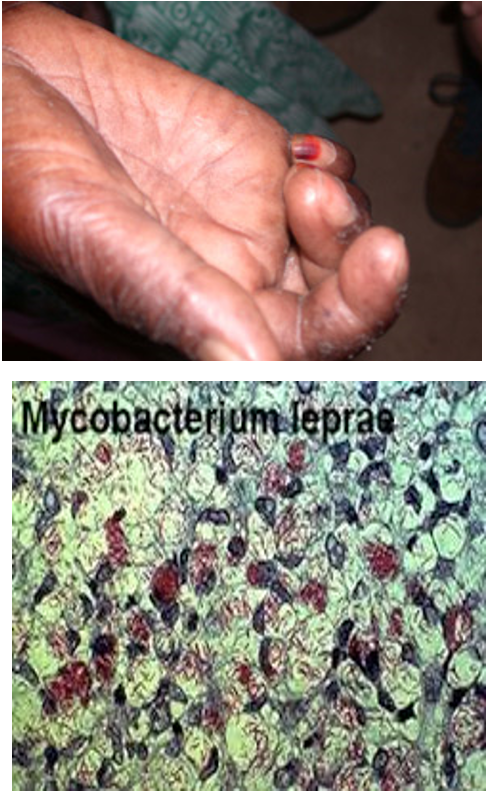
Maladie de Hansen (Lèpre tuberculoïde)
- Seconde mycobactériose mondiale après la tuberculose.
- Des zones de forte endémicité persistent. La maladie n'existe plus de façon autochtone en France métropolitaine, mais de nouveaux cas, originaires des DOM-TOM ou importés, sont régulièrement diagnostiqués.
- Tâches de dépigmentation
- Mononeuropathie multiple avec
- Hypo à Anesthésie
- Maux perforants, mutilations
- Hypertrophie des nerfs
- Diagnostic : mise en évidence du Bacille de Hansen (Mycobacterium leprae) >> coloration de Ziehl-Neelsen sur prélèvement de muqueuse, nerf,…
On la dit en coup de sabre
Patiente de 18 ans vue 6 mois après la survenue d’une sensation de douleur de l’hémicorps droit ayant duré plusieurs jours révélant un accident ischémique cérébral gauche profond et des micro-saignements sous-corticaux. Toutes les lésions cérébrales sont latéralisées au niveau de l’hémisphère gauche
Que vous évoque les anomalies cutanées sur cette photo ?
Lésions cutanées au niveau du front à gauche avec un aspect « en coup de sabre » fait dans ce cas de lésions hyper-pigmentées. Dans d’autres cas les lésions peuvent être hypo-pigmentées.
Il s’agit d’une sclérodermie « en coup de sabre »
Les lésions cutanées sont situées le plus souvent au niveau du front, habituellement unilatérales, sont associées à une ligne cicatricielle séparant le tissu atrophique du tissu normal.
L’évolution de la maladie est lente et progressive et se stabilise généralement aux alentours de 20 ans
L’atteinte du SNC est rare. Les lésions à l’IRM sont décrites comme étant du même côté que les lésions faciales
Le mécanisme physiopathologique des lésions cérébrales est mal connu et semble être une atteinte de type vascularite
Le traitement dans ce cas consiste en un traitement immunosuppresseur, par exemple le Méthotrexate
Caen 2020 interne
Un homme de 31 ans consulte aux urgences pour l’apparition la veille dans l’après-midi d’une monoparésie du membre supérieur droit. Ses antécédents se limitent à plusieurs traumatismes et à un tabagisme actif chiffré à environ 4 paquets-année. Alors qu’il effectuait une randonnée depuis 5 jours avec port de sac à dos d’environ 15kg, il décrit la survenue soudaine d’une douleur du creux axillaire droit à type de brûlure. La douleur bien soulagée par la prise d’un AINS est suivie 1/2h plus tard d’un déficit moteur du bras droit accompagné de quelques paresthésies de l’ensemble des doigts de la main droite. Il n’est pas rapporté de traumatisme récent de l’épaule, d’épisodes infectieux, de vaccination, de chirurgie récente ou d’altération de l’état général. Il est apyrétique. A l’examen neurologique, il est noté une amyotrophie du deltoïde à droite ; un déficit moteur à droite chiffré à 0/5 pour le deltoïde, les supra et infra-épineux, le biceps brachial et le triceps brachial, à 2/5 pour les fléchisseur/releveur du poignet et l’abducteur du pouce, et à 4/5 pour les interosseux ; une hypoesthésie tactile de la face externe du bras et de l’avant-bras droits ; des réflexes ostéo-tendineux tous abolis au membre supérieur droit, faibles au membre supérieur gauche, et normaux et symétriques aux membres inférieurs. L’examen des paires crâniennes est normal.
Quel est votre diagnostic et quel examen complémentaire demandez-vous ?
Il s’agit d’un syndrome de Parsonage-Turner ou névralgie amyotrophique. L’électroneuromyogramme chez ce patient a montré (1) en stimulodétection des potentiels sensitifs et moteurs normaux sur l'ensemble des nerfs explorés du membre supérieur droit jusqu’au point d’Erb et des ondes F présentes et normales sur l'ensemble des nerfs explorés, (2) en détection radiculaire de C5 à C8-D1, des tracés globalement neurogènes (appauvris accélérés voire simples) sur l'ensemble des muscles explorés, deltoïde (C5), biceps (C6), triceps et extenseur commun des doigts (C7), et de manière moins marquée premier interosseux, court abducteur du pouce et abducteur du V (C8-D1). Pas d'activité de repos relevée.
La douleur de l’épaule à type de brûlure, l’amyotrophie du deltoïde et le déficit moteur prédominant au plexus brachial chez un homme jeune sont des éléments orientant vers le diagnostic. Dans le cas présent, la disparition très rapide de la douleur est inhabituelle, tout comme l’apparition très rapide du déficit moteur (en général entre quelques heures et plusieurs jours après la douleur). En outre, les signes de dénervation à l’ENMG peuvent n’apparaître que jusqu’à 4 semaines après le début des symptômes. L’imagerie en IRM peut apporter des éléments de confirmation du diagnostic. La prise en charge habituelle repose actuellement sur la rééducation, indispensable afin d’éviter les complications musculaires et tendineuses, même si la plupart des patients récupère spontanément (principalement dans les 6 premiers mois). Si besoin, la corticothérapie à visée antalgique permet d’accélérer la phase de récupération.
Références :
- Gstoettner C et al., J Neurol Neurosurg Psychiatry 2020 ; 91: 879-888. doi:10.1136/jnnp-2020-323164
- Cruz-Martinez et al., J Peripher Nerv Syst 2002 ; 7(3): 198-204. doi: 10.1046/j.1529-8027.2002.02025.x.
No more laughing
Vous vous occupez d’un homme de 19 ans sans antécédent qui a été hospitalisé dans votre service pour l’apparition rapidement progressive sur 48h d’une instabilité à la marche. Il s’y associe des paresthésies symétriques des mains et des pieds d’aggravation progressive. Le patient ne se plaint pas de troubles moteurs ou sphinctériens.
Il n’y a pas d’infection récente ni d’autre plainte sur le plan général. Lorsque vous lui demandez s’il a consommé des toxiques, il vous rapporte avoir consommé 60 cartouches de protoxyde d’azote sur les 3 derniers jours à des fins récréatives.
Votre examen objective une ataxie proprioceptive majeure et des réflexes cutanéo plantaires en extension. Le reste de votre examen clinique est normal.
Vous faites réaliser l’IRM médullaire suivante :
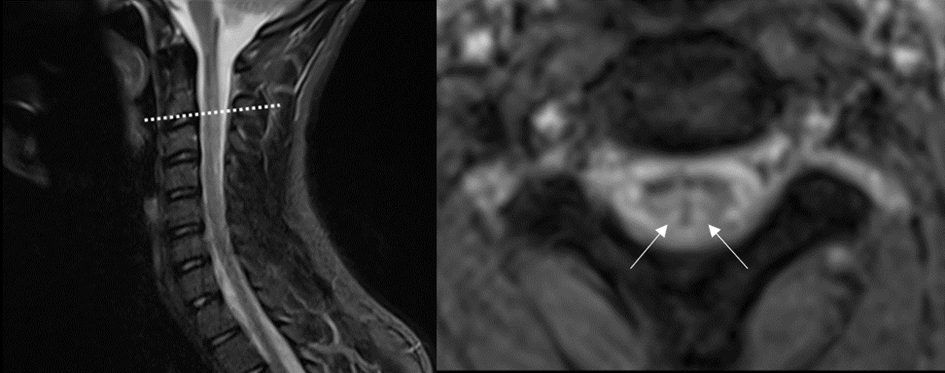
Les résultats biologiques mettent en évidence une macrocytose (109.7fL ; N=80-100), une augmentation de l’homocystéine (69.8µmol/L ; N=3,7-13,9), une élévation de la vitamine B12 (11.63µg/L ; N=0,22-0,91) et une augmentation de l’acide méthylmalonique (1.9µmol/L ; N<0.5).
Quel diagnostic évoquez-vous ?
Il s’agit d’une sclérose combinée de la moelle par déficit fonctionnel en vitamine B12 à la suite d’une intoxication au protoxyde d’azote (NO) (Noh et al. 2020).
Le protoxyde d’azote induit une oxydation irréversible des ions cobalt de la vitamine B12 à l’origine de son inactivation. La B12 participe à la transformation de l’homocystéine en méthionine et l’acide méthylmalonique en succinylCoA (Keddie et al. 2018). Ceci explique l’augmentation de la concentration en B12, en homocystéine et en acide méthylmalonique chez le patient.
La méthionine étant un élément important pour la méthylation des protéines de la myéline, sa diminution pourrait être à l’origine d’une démyélinisation, ici induite par le NO (Smith et al. 2006).
Le traitement repose sur la supplémentation en vitamine B12. Il n’y a cependant pas de recommandation concernant la posologie et la durée de traitement dans le cadre d’une intoxication par le NO. Chez le patient, la supplémentation a permis une correction des anomalies biologiques et une récupération complète de la symptomatologie en quelques mois.
La consommation de NO à visée récréative est en augmentation. Il s’agit donc d’une cause de sclérose combinée de la moelle à connaitre d’autant plus que le pronostic est souvent favorable après supplémentation (Noh et al. 2020).
Références
Keddie S, Adams A, Kelso ARC, Turner B, Schmierer K, Gnanapavan S, Malaspina A, Giovannoni G, Basnett I, Noyce AJ. No laughing matter: subacute degeneration of the spinal cord due to nitrous oxide inhalation. J Neurol. 2018 May;265(5):1089-1095. doi: 10.1007/s00415-018-8801-3. Epub 2018 Mar 3. PMID: 29502317; PMCID: PMC5937900.
Smith SE, Kinney HC, Swoboda KJ, Levy HL. Subacute combined degeneration of the spinal cord in cblC disorder despite treatment with B12. Mol Genet Metab. 2006 Jun;88(2):138-45. doi: 10.1016/j.ymgme.2006.02.007. Epub 2006 Mar 30. PMID: 16574454.
Noh T, Osman G, Chedid M, Hefzy H. Nitrous oxide-induced demyelination: Clinical presentation, diagnosis and treatment recommendations. J Neurol Sci. 2020 Jul 15;414:116817. doi: 10.1016/j.jns.2020.116817. Epub 2020 Apr 3. PMID: 32302804.
Collet serré
Une femme de 57 ans aux antécédents de tabagisme actif et d’emphysème pulmonaire vous est adressée pour l’apparition brutale de troubles phasiques associés à une cervicalgie intense il y a 24 heures.
Danseuse de salon occasionnelle, elle rapporte avoir assisté à un cours particulièrement dynamique la veille.
L’examen clinique retrouve une aphasie non fluente associée à une discrète hémiparésie brachio faciale droite. Le score NIHSS est évalué à 4.
Les résultats de l’IRM cérébrale et de l’angioscanner des troncs supra aortiques vous sont présentés ci dessous.
Qu’en pensez-vous ? Quel diagnostic étiologique retenez-vous ?
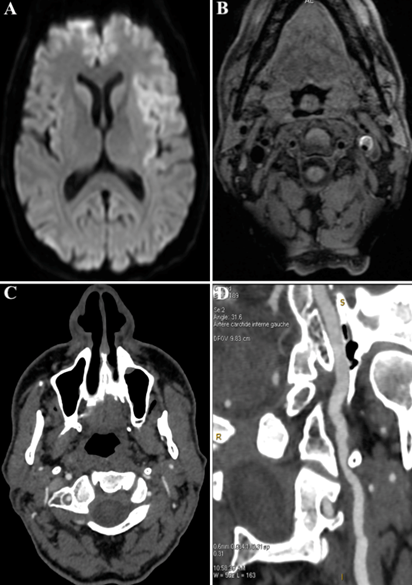
Il s’agit d’un infarctus cérébral sylvien superficiel gauche secondaire à une dissection de l’artère carotide interne gauche sous pétreuse au contact d’une apophyse styloïde trop longue (correspondant à un « syndrome d’Eagle »).
En effet, l’IRM cérébrale retrouve la présence d’un hypersignal diffusion intéressant le territoire sylvien superficiel gauche en faveur d’un infarctus cérébral récent (A). La séquence T1 FAT-SAT montre un hématome de paroi au niveau de l’artère carotide interne gauche sous pétreuse correspondant à une dissection aigue (B). L’angioscanner des TSA en coupe axiale retrouve une apophyse styloïde gauche venant au contact direct de l’artère carotide interne gauche disséquée (C). L’empreinte de l’apophyse styloïde gauche sur l’artère carotide interne est bien visible sur les coupes axiales et sagittales (C, D).
Les dissections carotidiennes représentent une cause importante d’infarctus cérébral du sujet jeune. On les classe classiquement en « spontanées» ou « traumatiques» même si le traumatisme en question est parfois minime (1).
Toutefois la dissection peut aussi être favorisée par la survenue de microtraumatismes répétés en raison de la proximité entre une artère et une structure osseuse de voisinage. L’apophyse styloïde est une structure osseuse qui se projette inférieurement, antérieurement et médialement à partir de la surface inférieure de l’os temporal. Sa longueur normale est inférieure à 25 mm.
Une apophyse styloïde trop longue est retrouvée chez environ 4% des individus notamment en cas de calcifications du ligament stylo hyoïde et peut, dans certains cas dépasser 75 mm (2).
Le syndrome d’Eagle porte le nom de celui l’ayant décrit pour la 1ère fois en 1937 et regroupe deux présentations cliniques distinctes. Le syndrome classique qui correspond à l’ensemble des symptômes ORL en lien avec la présence d’une apophyse styloïde trop longue (douleur orophrayngée unilatérale irradiant vers l’oreille, irritation mécanique de certaines paires crâniennes notamment le nerf glossopharyngien).
Le syndrome d’Eagle vasculaire qui survient lorsque l’apophyse styloïde trop longue comprime l’artère carotide interne ou externe avec laquelle elle est en contact, favorisant ainsi la survenue de phénomènes hémodynamiques ou de dissections artérielles (3). Chez notre patient l’apophyse styloïde gauche est mesurée à 33 mm.
Un syndrome d’Eagle vasculaire peut aussi être occasionné par une apophyse styloïde de taille normale mais dont l’angulation particulière la met en contact avec l’artère carotide interne, en particulier lorsque la tête du patient n’est pas en position neutre (4). La réalisation de clichés dynamiques est alors particulièrement utile.
Le traitement n’est pas clairement codifié mais une styloidectomie unilatérale peut se discuter dans certains cas.
Les reconstructions 3D à partir de l’angioscanner des TSA confirment la présence d’une apophyse styloïde gauche trop longue venant en contact et comprimant l’artère carotide interne ipsilatérale.
1. Schievink WI. Spontaneous dissection of the carotid and vertebral arteries. N Engl J Med 2001 ; 344 :898-906
2. Eagle WW : Elongated styloid process : symptoms and treatment. AMA Arch Otolaryngol 67 : 172-176, 1958
3. Eagle WW : Symptomatic elongated styloid process : report of two cases of styloid process-carotid artery syndrome with operation. Arch Otolaryngol 49 :490-503, 1949
4. Chuang WC, Short JH, Mc Kinney AM et al. Reversible left hemispheric ischemia secondary to carotid compression in Eagle syndrom :surgical and CT angiographic correlation.
Une histoire à vomir couché !
On vous appelle aux urgences pédiatriques pour discuter d’un cas.
Maeva à 6 ans, est née à terme, n’a pas antécédents particuliers avec un développement psychomoteur normal. Elle est au CP et tout se déroule sans incident.
C’est la 4ème fois que ses parents l’amènent aux urgences pour des épisodes de malaises. Ces derniers ont lieu plutôt la nuit. La maman vous raconte que Maeva se réveille et l’appelle. Elle retrouve sa fille dans son lit, pâle, confuse. Elle vomit à chaque fois. Le dernier malaise était plus inquiétant avec une enfant inconsciente et « molle comme une poupée de chiffon ».
Aux urgences, l’examen clinique est normal. Le bilan biologique standard ne retrouve aucune anomalie. Les examens d’imagerie abdominale sont normaux. Il a été conclu à des épisodes viraux gastro-intestinaux et une consultation de gastro-entérologie est programmée. Cette hypothèse ne vous séduit pas et notamment du fait du dernier malaise. Un diagnostic vous vient alors en tête et pour le confirmer, vous faites réaliser l’EEG présenté ci-dessous. Quel est alors votre diagnostic ?
Syndrome de Panayiotopopoulos
Le syndrome de Panayiotopoulos ou épilepsie occipitale bénigne à début précoce débute entre 1 et 14 ans (6 ans en moyenne). Le sexe ratio est de 1:1. Ses manifestations représentent 7 à 13% des épilepsies focales pédiatriques.
Les crises sont nocturnes dans 2/3 des cas et se manifestant principalement par des symptômes neurovégétatifs. Les crises se caractérisent par des sensations de nausée et fréquemment de vomissements. D’autres symptômes dysautonomiques peuvent être observés comme une pâleur, une mydriase, des altérations cardio-respiratoires et thermorégulatrices. Les crises peuvent également s’accompagner de mouvements de déviation des yeux et évoluer vers des clonies hémi corporelles voire une généralisation secondaire.
Les principaux diagnostics différentiels sont la migraine, une syncope, une gastro-entérite.
Les crises ont des durées variables, pouvant être brèves ou durer plusieurs heures
L’EEG montre classiquement des pointes ou ondes lentes occipitales, accentuées par le sommeil et l’hyperventilation et bloquées à l’ouverture des yeux.
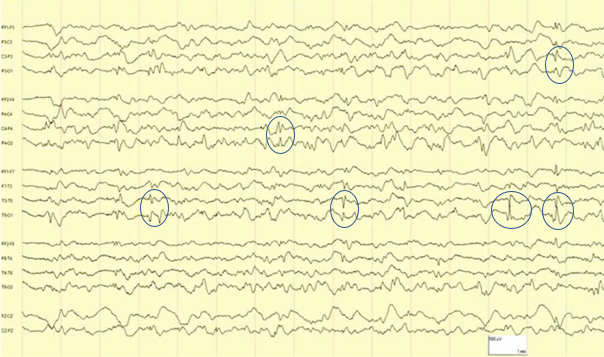
L’imagerie cérébrale est normale.
Concernant la thérapeutique, une abstention est possible si les crises sont rares. Un traitement antiépileptique ne se justifie que pour les enfants qui présentent des crises fréquentes, prolongées et qui perturbent significativement la qualité de vie de l’enfant.
Le pronostic est en général bon avec un arrêt des crises un à deux ans après le début de la maladie.
Bibliographie :
Parisi P, Villa MP, Pelliccia A, Rollo VC, Chiarelli F, Verrotti A. Panayiotopoulos syndrome: diagnosis and management. Neurol Sci. 2007 Apr;28(2):72-9. doi: 10.1007/s10072-007-0790-4. PMID: 17464469
Graziosi A, Pellegrino N, Di Stefano V, Raucci U, Luchetti A, Parisi P. Misdiagnosis and pitfalls in Panayiotopoulos syndrome. Epilepsy Behav. 2019 Sep;98(Pt A):124-128. doi: 10.1016/j.yebeh.2019.07.016. Epub 2019 Jul 29. PMID: 31369969.
The Tony Stark story
Vous voyez en consultation un homme de 36 ans. Il présente comme antécédents des convulsions hyperthermiques dans l’enfance, une sténose du pylore à 3 semaines de vie, une déficience intellectuelle avec retard développemental. Il habite dans un foyer de vie depuis l’adolescence. Le caryotype et l’IRM cérébrale réalisés à l’âge de 12 ans sont normaux.
Depuis un an, ce patient présente une perte progressive de la marche avec dégradation cognitivo-comportementale, apathie, hypotension orthostatique et une dyspraxie.
A l’examen, vous retrouvez une rigidité extrapyramidale de l’hémicorps gauche qui prédomine au membre supérieur ainsi que des rétractions tendineuses.
Voici une nouvelle IRM cérébrale réalisée quelques jours avant votre consultation.
Quel est votre diagnostic ?
BPAN (Beta propeller protein-associated neurodegeneration)
Il s’agit d’une BPAN pour Beta Propeller protein-Associated Neurodegeneration. Qui est une cause rare de neurodégénération avec accumulation de fer appartenant à la famille des NBIA (Neurodegeneration with Brain Iron Accumulation).
La BPAN est responsable d’un retard psychomoteur, d’une épilepsie, d’une atteinte pyramidale, d’un syndrome extra-pyramidal, d’une déficience intellectuelle et de troubles du comportement de type autistique. Le phénotype peut etre variable, mais l’évolution en deux temps (retard intellectuel dans l’enfance avec épilepsie puis développement de troubles moteurs neurologiques au début de l’âge adulte) doivent évoquer le diagnostic. L’apparition d’image typique à l’IRM cérébrale dans la seconde phase d’évolution, tel que des halos hyper-intenses et symétriques entourant une bande centrale hypo-intense dans la substance noire en séquence pondérée en T1 est pathognomonique des BPAN. On retrouve également sur les séquences T2 et SWI une hypo-intensité dans la substance noire et les pédoncules cérébraux correspondant aux zones de dépots accru de fer.
La transmission est dominante liée à l’X et le diagnostic est établi par l’identification d’une mutation du gène WDR45. Ceci dit, la grande majorité des cas sont des cas sporadiques résultant d’une mutation de novo.
L’IRM cérébrale permet d’évoquer le diagnostic et la mutation
A ce jour il n’y a pas de traitements curatifs des BPAN, mais les chélateurs de fer sont une piste en cours d’exploration.
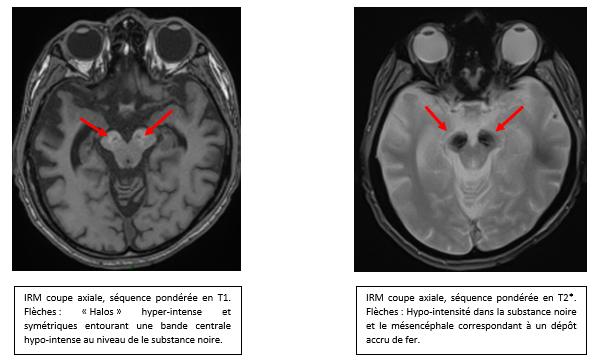
Bibliographie :
Amaral LLF, Gaddikeri S, Chapman PR, Roy R, Gaddikeri RS, Marussi VH, et al. Neurodegeneration with Brain Iron Accumulation: Clinicoradiological Approach to Diagnosis. J Neuroimaging Off J Am Soc Neuroimaging. août 2015;25(4):539‑51.
Hor CHH, Tang BL. Beta-propeller protein-associated neurodegeneration (BPAN) as a genetically simple model of multifaceted neuropathology resulting from defects in autophagy. Rev Neurosci. 24 avr 2019;30(3):26177.
Un train peut en cacher un autre
Neurologie en raison de mouvements anormaux de nature choréique apparus depuis quelques mois. IL n’y a aucun antécédant familial connu, mais la recherche de mutation du gène IT5 conduit au diagnostic de maladie de Huntington. Un traitement par Tétrabénazine 75 mg/J permet une amélioration significative des mouvements choréiques. Début 2014, le patient est revu pour une consultation de contrôle dans le cadre de sa maladie de Huntington. Il signale des troubles importants de la marche nécessitant depuis quelques mois l’utilisation d’un appui unilatéral.
L’examen neurologique ne note pas de mouvements choréiques. La marche est spastique avec un fauchage bilatéral. IL existe une instabilité bilatérale à la manœuvre de Mingazzini, les réflexes ostéotendineux sont vifs aux membres inférieurs et il existe un signe de Babinski bilatéral.
Une IRM médullaire est réalisée.
- Un bilan complémentaire est- il justifié dans ce contexte ? Si oui lequel ?
- Comment l’interprétez vous ? Que proposez vous ?
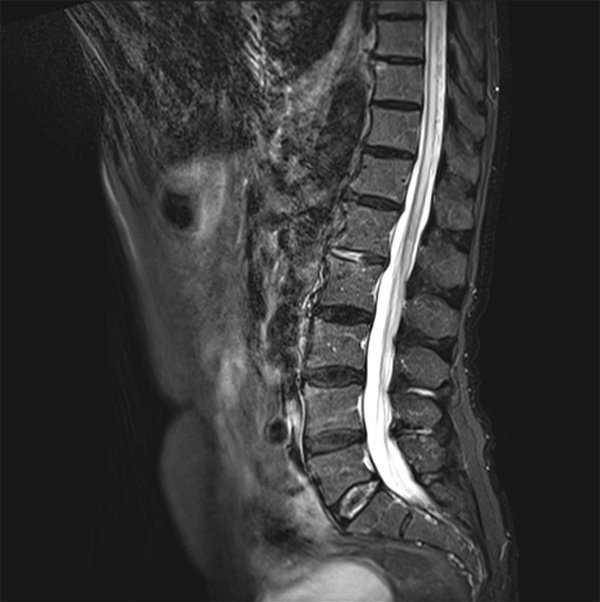
L’IRM médullaire montre un hypersignal intramédullaire étendu de T7 à D1 et des dilatations vasculaires postérieures au niveau du cône terminal en faveur d’une fistule durale.
Une artériographie médullaire est réalisée : l’injection de la 8ème artère intercostale droite opacifie des vaisseaux anormaux au niveau de la moelle épinière correspondant au drainage veineux périmédullaire de la fistule durale.
Une embolisation est programmée.
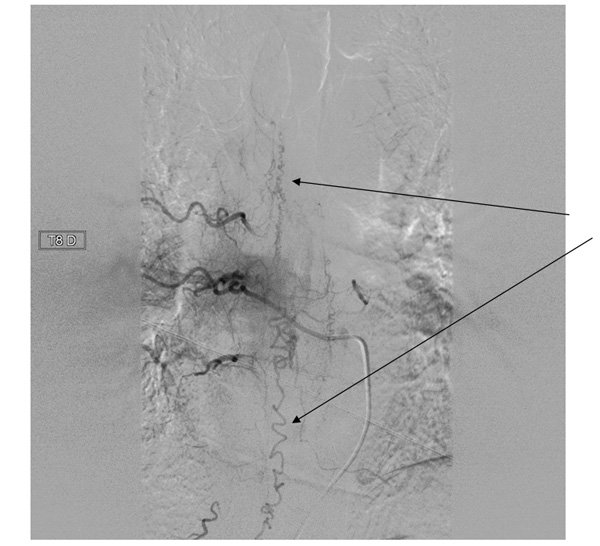
Les fistules durales rachidiennes correspondent à un shunt situé dans la dure mère, entre une artère méningée et le système veineux médullaire . Il s’agit de la plus fréquente malformation vasculaire médullaire. Elle survient préférentiellement chez l’homme entre 50 et 60 ans.
La souffrance médullaire est secondaire à une hypertension veineuse ou à des lésions d’ischémie veineuse. Cette souffrance se manifeste par des signes de claudication médullaire, un syndrome pyramidal, des troubles sphinctériens, des troubles proprioceptifs . L’installation des signes cliniques est souvent insidieuse, mais de rares cas de début brutal (myelopathie ischémique ou rarement hémorragie).
L’IRM médullaire montre le plus souvent au niveau du cône terminal médullaire, un hypersignal T2 médullaire étendu et un élargissement de la moelle (œdème médullaire), et des images serpigineuses correspondant à des veines périmédullaires dilatées (hypointenses en T2 se rehaussant après injection de gadolinium T1). Ces signes n’ont pas de valeur localisatrice de la fistule (qui peut être cervicale, thoracique, lombaire).
Une artériographie médullaire met en évidence la fistule dont le traitement est endovasculaire (embolisation)
Pour en savoir plus :
Hacein-Bey L, Konstas AA, Pile-Spellman J Natural history, current concepts, classification, factors impacting endovasculartherapy, and pathophysiology of cerebral and spinal dural arteriovenous fistulas Clin Neurol Neurosurg. 2014 Jun;121:64-75.
Jellema K, Tijssen CC, van Gijn J. Spinal dural arteriovenous fistulas: a congestive myelopathy that initially mimics a peripheral nerve disorder Brain. 2006 Dec;129(Pt 12):3150-64.
Yen PP, Ritchie KC, Shankar JJ. Spinal dural arteriovenous fistula: correlation between radiological and clinical findings. J Neurosurg Spine. 2014 Aug 15:1-6. [Epub ahead of print]
Une explosion dans la tête
C’est un homme de 30 ans qui consulte pour « un sentiment de déflagration dans la tête ».
Cette symptomatologie évolue depuis l’âge de 16-17 ans, à raison de 3 à 4 épisodes par an. C’est le soir, au moment de l’endormissement que surviennent ces épisodes qu’il décrit en regard du vertex, paracentral droit, discrètement en arrière, ne prenant pas toute la tête. Il les ressent comme « choquant, très bizarre et très surprenant». L’accès est bref, non associé à une gêne ou une douleur par la suite. Le réendormissement est plutôt facile.
Quel est votre diagnostic ?
- Syndrome de la tête qui explose
- Variante explosive du ice pick headache
- Céphalée hypnique
- Forme atypique de myoclonies d’endormissement
- Crise focale hypnique
Le syndrome de la tête qui explose ; exploding head syndrome
Le syndrome de la tête qui explose figure dans le chapitre « autres parasomnies » dans la classification internationale des troubles du sommeil (ICSD-III).
Il s’agit de la perception d’un bruit soudain ou l’impression d’une violente explosion dans la tête à l’endormissement ou lors d’un réveil nocturne. L’événement entraîne un réveil brutal souvent associé à un sentiment de peur. L’expérience n’est pas associée à des plaintes importantes de douleur et ne nécessite pas de traitement particulier.
Les mécanismes neurophysiologiques sous-jacents sont inconnus.
L’évolution est bénigne. En cas d’événements répétés dans la même nuit, le syndrome de la tête qui explose peut entraîner une insomnie. Le traitement repose alors sur une bonne hygiène du sommeil et une approche cognitivo-comportemental de l’insomnie.
American Academy of Sleep Medicine (AASM). The International classification of sleep disorders: diagnostic & coding manual (ICSD-3). 3rd edn. Darien, IL: American Academy of Sleep Medicine; 2014.
Exploding head syndrome: clinical features, theories about etiology, and prevention strategies in a large international sample. Sharpless BA, Denis D, Perach R, French CC, Gregory AM. Sleep Med. 2020 Nov;75:251-255. doi: 10.1016/j.sleep.2020.05.043. Epub 2020 Jun 10. PMID: 32862013
Exploding Head Syndrome: a Review. Ceriani CEJ, Nahas SJ. Curr Pain Headache Rep. 2018 Jul 30;22(10):63. doi: 10.1007/s11916-018-0717-1. PMID: 30062616
Pour en savoir plus :
1.Bassi SS, Bulundwe KK, Greeff GP, Labuscagne JH, Gledhill RF. MRI of the spinal cord in myelopathy complicating vitamin B12 deficiency: two additional cases and a review of the literature. Neuroradiology 1999;41:271-274.
2.Kumar N. Metabolic and toxic myelopathies. Semin Neurol 2012;32:123-136.
3.Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med 2006;21:1063-1068.
4.Berger JR, Quencer R. Reversible myelopathy with pernicious anemia: clinical/MR correlation. Neurology 1991;41:947-948.
5.Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract 2012;97:359-367.
6. P. Coignard, M. Lemesle, G. Madinier, E. Manceau, G. Osseby, B. Lucas, N. Baudouin, D. Martin, M. Giroud, R. Dumas. Intérêt de l'imagerie par résonance magnétique dans la sclérose combinée de la moelle par carence en vitamine B12. La revue neurologique 2000. Page : 1000