Du rire aux larmes
Vous voyez en consultation Mme G. âgée de 49 ans amenée par son époux pour des troubles psycho-comportementaux.
Elle n’a pas d’antécédent personnel. Vous notez une fin de vie difficile pour son père avec troubles psycho comportementaux alors qu’il était en fauteuil roulant (on aurait parlé de la maladie de Charcot). Son frère plus âgé aurait des difficultés à marcher et serait en cours de bilan neurologique.
Mme G. vous regarde avec un grand sourire et vous explique que tout va bien. Son mari raconte qu’il vit un enfer depuis plusieurs mois. Son épouse institutrice en maternelle n’arrive plus à gérer sa classe. Elle ne parvient pas à organiser l’enseignement et à même frappé des enfants si bien que des demandes de confrontation sont en cours avec des parents. A la maison, le mari note de nombreux oublis, des passages de coq à l’âne, une impatience importante nouvelle (elle ne peut plus coudre ou lire car se désintéresse vite). Ses amis se sont éloignés car elle est devenue plus franche avec de nombreuses remarques déplacées. Le mari note aussi une négligence physique puisqu’elle ne se maquille plus et ne change que très rarement ses habits. Il décrit aussi une prise de 10 kg sur un an car elle mange sans cesse des gâteaux alors qu’elle mangeait peu de sucré par le passé. L’autonomie est grandement altérée.
L’examen clinique ne retrouve pas d’anomalie.
L’examen neuropsychologique retrouve un syndrome dysexécutif comportemental et cognitif, un ludisme important et beaucoup de triche durant les passations. La mémoire est conservée, il n’y a pas de trouble instrumental. Elle n’a malheureusement pas pu être explorée sur le plan orthophonique.
Voici une IRM cérébrale et un PET-Scanner.
Que dire de l’imagerie ? Quel est le diagnostic neuropsychologique ? Quel diagnostic plus général peut-on proposer ?
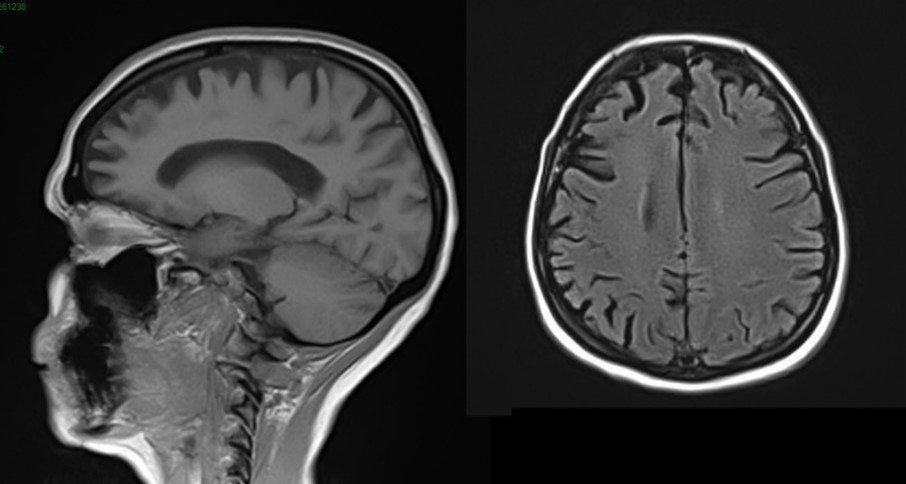
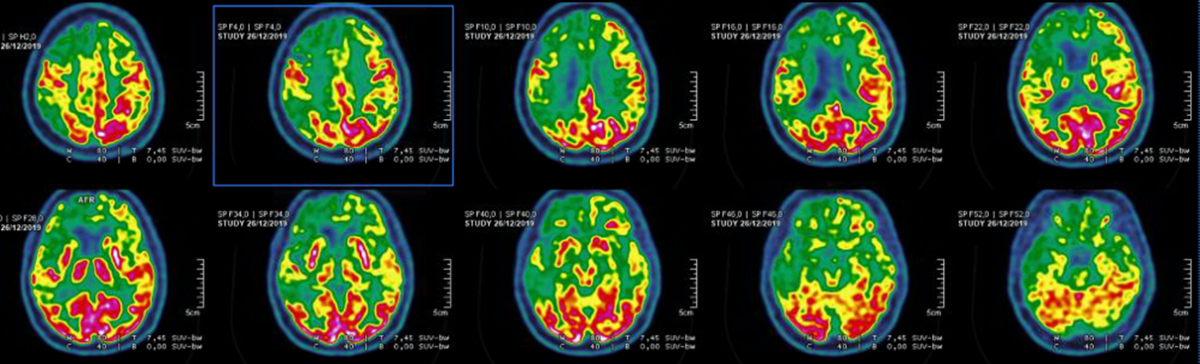
Atrophie frontale sur l’IRM avec hypométabolisme antérieur marqué, démence fronto-temporale, tableau de mutation C9ORF72
La démence frontotemporale (DFT) fait partie du groupe des dégénérescences lobaires fronto temporales (DFT, DCB, PSP, APP non fluente et sémantique). Ici, le tableau clinique remplit les critères cliniques de DFT possible selon Rascovsky, soit au moins 3 critères parmi :
Désinhibition comportementale précoce,
Apathie/inertie précoce,
Perte de sympathie ou d’empathie précoce,
Comportement persévératif, stéréotypé ou compulsif/obsessionnel précoce,
Hyperoralité et changement des habitudes alimentaires,
Profil neuropsychologique (déficit exécutif, préservation mémoire épisodique et fonctions visuo spatiales).
Le diagnostic devient probable car aux critères cliniques de DFT possible s’ajoute un déclin fonctionnel significatif et des résultats d’imageries concordants (IRM et PET).
L’histoire familiale permettra probablement de passer en stade certain puisqu’elle semble orienter vers une mutation C9ORF72.
On estime à 40% l’origine génétique des DFT. Cela motive la recherche systématique d’une histoire familiale. La mutation C9ORF72 est finalement de description récente mais probablement la plus fréquente. Elle doit systématiquement être évoquée dans une histoire familiale mêlant DFT et SLA.
Une histoire renversante
Vous voyez en consultation un patient âgé de 66 ans. Il a comme antécédents une agression physique en 2003 avec un état de stress post traumatique séquellaire et une toux chronique depuis 10 ans avec bilan étiologique pneumologique et gastro-entérologique négatif.
Depuis l’agression, il décrit l’installation progressive de troubles de la marche avec chutes itératives parfois traumatiques.
Votre examen clinique retrouve une marche ataxique avec élargissement du polygone de sustentation, un Romberg positif, une marche en funambule impossible et aucun déficit moteur. Les réflexes tendineux ne sont pas perçus. La pallesthésie est abolie aux membres inférieurs jusqu’aux genoux et aux membres supérieurs jusqu’aux coudes.
L’étude des vitesses de conduction motrices est normale comme l’examen de détection à l’aiguille. L’étude des vitesses de conduction sensitives vous est proposé ci-dessous. Il n’y a aucune réponse reproductible aux quatre membres lors de l’enregistrement des potentiels évoqués somesthésiques. Une IRM cérébrale est réalisée. Une coupe sagittale pondérée en T1 est proposée ci-dessous.
Quel est votre diagnostic ?
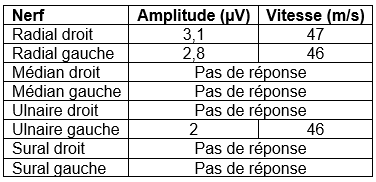
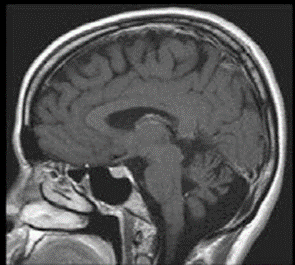
Syndrome de CANVAS
Il s’agit d’un syndrome de CANVAS (Cerebellar Ataxia Neuropathy Vestibular Areflexia Syndrome) caractérisé par la triade trouble cérébelleux, atteinte vestibulaire bilatérale et déficit sensitif. L’atteinte neurologique est d’évolution lente et d’apparition tardive à l’âge adulte. On peut également retrouver dans presque deux-tiers des cas une toux chronique et dans un tiers des cas des symptômes dysautonomiques.
En général les potentiels évoqués somesthésiques sont absents et l’ENMG montre un aspect de ganglionopathie. L’IRM encéphalique retrouve une atrophie cérébelleuse et l’examen vestibulaire une anomalie du réflexe visio-oculo-vestibulaire (aréflexie vestibulaire bilatérale assez caractéristique).
L’apparition d’un syndrome de CANVAS est souvent tardive et sporadique. Le diagnostic nécessite d’éliminer les diagnostics différentiels tel que le SCA3 ou la maladie de Friedreich. Des cas familiaux ont été décrit, faisant découvrir une répétition intronique AAGGG récessive dans le gène RFC1.
Bibliographie :
Szmulewicz DJ, McLean CA, MacDougall HG, Roberts L, Storey E, Halmagyi GM. CANVAS an update: clinical presentation, investigation and management. J Vestib Res Equilib Orientat. 2014;24(56):465‑74.
Szmulewicz DJ, Roberts L, McLean CA, MacDougall HG, Halmagyi GM, Storey E. Proposed diagnostic criteria for cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Neurol Clin Pract. févr 2016;6(1):61‑8.
Peillet C, Buch D, Nifle C, Servan J, Pico F. Ataxie cérébelleuse avec neuropathie et aréflexie vestibulaire bilatérale : le syndrome CANVAS, démarche diagnostique. Prat Neurol - FMC. déc 2018;9(4):268‑71.
Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yau WY, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet. avr 2019;51(4):649‑58.
Cortese A, Reilly MM, Houlden H. RFC1 CANVAS / Spectrum Disorder. 2020
Nov 25. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A, editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993–2021. PMID: 33237689.
15/06/2023
admin
15/06/2023
admin
15/06/2023
admin
15/06/2023
admin
15/06/2023
admin
15/06/2023
admin
15/06/2023
admin
15/06/2023
admin
Bibliographie :
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. >Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain [Internet] 2011;134(9):2456–2477
De Jesus-Hernandez M.Mackenzie IR, Boeve BF et al.Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72(2):245-56.
Renton AE, Majounie E, Waite A et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72(2):257-68.