No more laughing
Vous vous occupez d’un homme de 19 ans sans antécédent qui a été hospitalisé dans votre service pour l’apparition rapidement progressive sur 48h d’une instabilité à la marche. Il s’y associe des paresthésies symétriques des mains et des pieds d’aggravation progressive. Le patient ne se plaint pas de troubles moteurs ou sphinctériens.
Il n’y a pas d’infection récente ni d’autre plainte sur le plan général. Lorsque vous lui demandez s’il a consommé des toxiques, il vous rapporte avoir consommé 60 cartouches de protoxyde d’azote sur les 3 derniers jours à des fins récréatives.
Votre examen objective une ataxie proprioceptive majeure et des réflexes cutanéo plantaires en extension. Le reste de votre examen clinique est normal.
Vous faites réaliser l’IRM médullaire suivante :
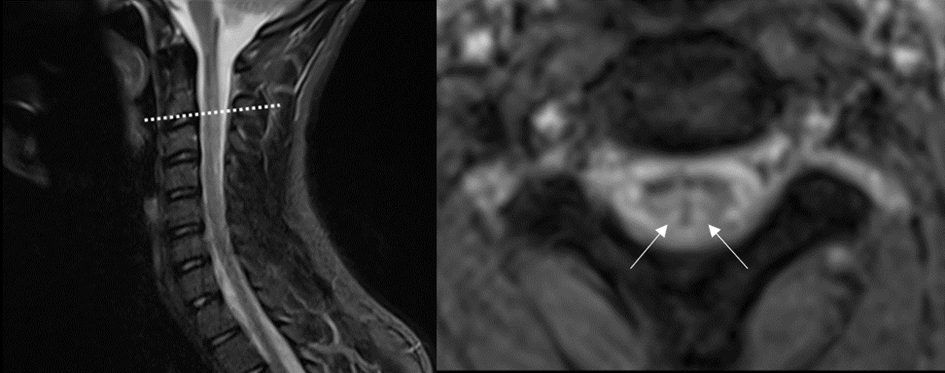
Les résultats biologiques mettent en évidence une macrocytose (109.7fL ; N=80-100), une augmentation de l’homocystéine (69.8µmol/L ; N=3,7-13,9), une élévation de la vitamine B12 (11.63µg/L ; N=0,22-0,91) et une augmentation de l’acide méthylmalonique (1.9µmol/L ; N<0.5).
Quel diagnostic évoquez-vous ?
Il s’agit d’une sclérose combinée de la moelle par déficit fonctionnel en vitamine B12 à la suite d’une intoxication au protoxyde d’azote (NO) (Noh et al. 2020).
Le protoxyde d’azote induit une oxydation irréversible des ions cobalt de la vitamine B12 à l’origine de son inactivation. La B12 participe à la transformation de l’homocystéine en méthionine et l’acide méthylmalonique en succinylCoA (Keddie et al. 2018). Ceci explique l’augmentation de la concentration en B12, en homocystéine et en acide méthylmalonique chez le patient.
La méthionine étant un élément important pour la méthylation des protéines de la myéline, sa diminution pourrait être à l’origine d’une démyélinisation, ici induite par le NO (Smith et al. 2006).
Le traitement repose sur la supplémentation en vitamine B12. Il n’y a cependant pas de recommandation concernant la posologie et la durée de traitement dans le cadre d’une intoxication par le NO. Chez le patient, la supplémentation a permis une correction des anomalies biologiques et une récupération complète de la symptomatologie en quelques mois.
La consommation de NO à visée récréative est en augmentation. Il s’agit donc d’une cause de sclérose combinée de la moelle à connaitre d’autant plus que le pronostic est souvent favorable après supplémentation (Noh et al. 2020).
Collet serré
Une femme de 57 ans aux antécédents de tabagisme actif et d’emphysème pulmonaire vous est adressée pour l’apparition brutale de troubles phasiques associés à une cervicalgie intense il y a 24 heures.
Danseuse de salon occasionnelle, elle rapporte avoir assisté à un cours particulièrement dynamique la veille.
L’examen clinique retrouve une aphasie non fluente associée à une discrète hémiparésie brachio faciale droite. Le score NIHSS est évalué à 4.
Les résultats de l’IRM cérébrale et de l’angioscanner des troncs supra aortiques vous sont présentés ci dessous.
Qu’en pensez-vous ? Quel diagnostic étiologique retenez-vous ?
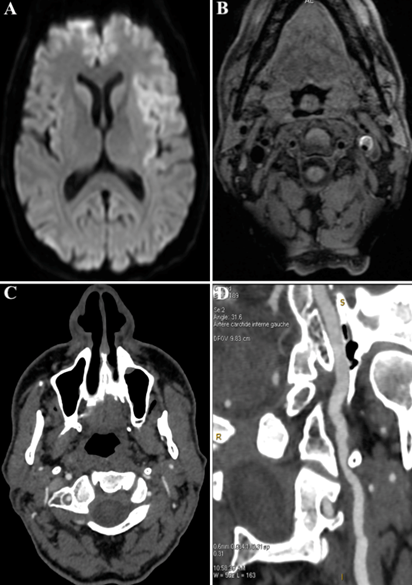
Il s’agit d’un infarctus cérébral sylvien superficiel gauche secondaire à une dissection de l’artère carotide interne gauche sous pétreuse au contact d’une apophyse styloïde trop longue (correspondant à un « syndrome d’Eagle »).
En effet, l’IRM cérébrale retrouve la présence d’un hypersignal diffusion intéressant le territoire sylvien superficiel gauche en faveur d’un infarctus cérébral récent (A). La séquence T1 FAT-SAT montre un hématome de paroi au niveau de l’artère carotide interne gauche sous pétreuse correspondant à une dissection aigue (B). L’angioscanner des TSA en coupe axiale retrouve une apophyse styloïde gauche venant au contact direct de l’artère carotide interne gauche disséquée (C). L’empreinte de l’apophyse styloïde gauche sur l’artère carotide interne est bien visible sur les coupes axiales et sagittales (C, D).
Les dissections carotidiennes représentent une cause importante d’infarctus cérébral du sujet jeune. On les classe classiquement en « spontanées» ou « traumatiques» même si le traumatisme en question est parfois minime (1).
Toutefois la dissection peut aussi être favorisée par la survenue de microtraumatismes répétés en raison de la proximité entre une artère et une structure osseuse de voisinage. L’apophyse styloïde est une structure osseuse qui se projette inférieurement, antérieurement et médialement à partir de la surface inférieure de l’os temporal. Sa longueur normale est inférieure à 25 mm.
Une apophyse styloïde trop longue est retrouvée chez environ 4% des individus notamment en cas de calcifications du ligament stylo hyoïde et peut, dans certains cas dépasser 75 mm (2).
Le syndrome d’Eagle porte le nom de celui l’ayant décrit pour la 1ère fois en 1937 et regroupe deux présentations cliniques distinctes. Le syndrome classique qui correspond à l’ensemble des symptômes ORL en lien avec la présence d’une apophyse styloïde trop longue (douleur orophrayngée unilatérale irradiant vers l’oreille, irritation mécanique de certaines paires crâniennes notamment le nerf glossopharyngien).
Le syndrome d’Eagle vasculaire qui survient lorsque l’apophyse styloïde trop longue comprime l’artère carotide interne ou externe avec laquelle elle est en contact, favorisant ainsi la survenue de phénomènes hémodynamiques ou de dissections artérielles (3). Chez notre patient l’apophyse styloïde gauche est mesurée à 33 mm.
Un syndrome d’Eagle vasculaire peut aussi être occasionné par une apophyse styloïde de taille normale mais dont l’angulation particulière la met en contact avec l’artère carotide interne, en particulier lorsque la tête du patient n’est pas en position neutre (4). La réalisation de clichés dynamiques est alors particulièrement utile.
Le traitement n’est pas clairement codifié mais une styloidectomie unilatérale peut se discuter dans certains cas.
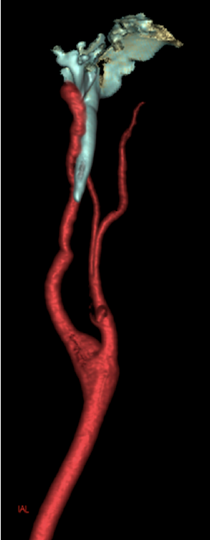
Les reconstructions 3D à partir de l’angioscanner des TSA confirment la présence d’une apophyse styloïde gauche trop longue venant en contact et comprimant l’artère carotide interne ipsilatérale.
1. Schievink WI. Spontaneous dissection of the carotid and vertebral arteries. N Engl J Med 2001 ; 344 :898-906
2. Eagle WW : Elongated styloid process : symptoms and treatment. AMA Arch Otolaryngol 67 : 172-176, 1958
3. Eagle WW : Symptomatic elongated styloid process : report of two cases of styloid process-carotid artery syndrome with operation. Arch Otolaryngol 49 :490-503, 1949
4. Chuang WC, Short JH, Mc Kinney AM et al. Reversible left hemispheric ischemia secondary to carotid compression in Eagle syndrom :surgical and CT angiographic correlation.
Une histoire à vomir couché !
On vous appelle aux urgences pédiatriques pour discuter d’un cas.
Maeva à 6 ans, est née à terme, n’a pas antécédents particuliers avec un développement psychomoteur normal. Elle est au CP et tout se déroule sans incident.
C’est la 4ème fois que ses parents l’amènent aux urgences pour des épisodes de malaises. Ces derniers ont lieu plutôt la nuit. La maman vous raconte que Maeva se réveille et l’appelle. Elle retrouve sa fille dans son lit, pâle, confuse. Elle vomit à chaque fois. Le dernier malaise était plus inquiétant avec une enfant inconsciente et « molle comme une poupée de chiffon ».
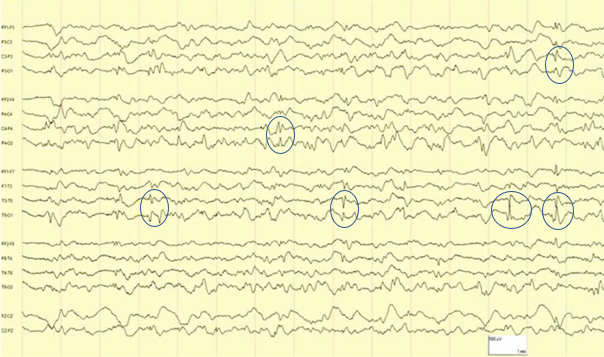
Aux urgences, l’examen clinique est normal. Le bilan biologique standard ne retrouve aucune anomalie. Les examens d’imagerie abdominale sont normaux. Il a été conclu à des épisodes viraux gastro-intestinaux et une consultation de gastro-entérologie est programmée. Cette hypothèse ne vous séduit pas et notamment du fait du dernier malaise. Un diagnostic vous vient alors en tête et pour le confirmer, vous faites réaliser l’EEG présenté ci-dessous. Quel est alors votre diagnostic ?
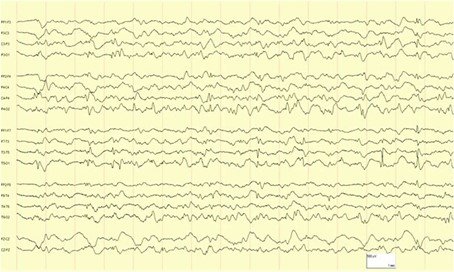
Syndrome de Panayiotopopoulos
Le syndrome de Panayiotopoulos ou épilepsie occipitale bénigne à début précoce débute entre 1 et 14 ans (6 ans en moyenne). Le sexe ratio est de 1:1. Ses manifestations représentent 7 à 13% des épilepsies focales pédiatriques.
Les crises sont nocturnes dans 2/3 des cas et se manifestant principalement par des symptômes neurovégétatifs. Les crises se caractérisent par des sensations de nausée et fréquemment de vomissements. D’autres symptômes dysautonomiques peuvent être observés comme une pâleur, une mydriase, des altérations cardio-respiratoires et thermorégulatrices. Les crises peuvent également s’accompagner de mouvements de déviation des yeux et évoluer vers des clonies hémi corporelles voire une généralisation secondaire.
Les principaux diagnostics différentiels sont la migraine, une syncope, une gastro-entérite.
Les crises ont des durées variables, pouvant être brèves ou durer plusieurs heures
L’EEG montre classiquement des pointes ou ondes lentes occipitales, accentuées par le sommeil et l’hyperventilation et bloquées à l’ouverture des yeux.
L’imagerie cérébrale est normale.
Concernant la thérapeutique, une abstention est possible si les crises sont rares. Un traitement antiépileptique ne se justifie que pour les enfants qui présentent des crises fréquentes, prolongées et qui perturbent significativement la qualité de vie de l’enfant.
Le pronostic est en général bon avec un arrêt des crises un à deux ans après le début de la maladie.
Bibliographie :
Parisi P, Villa MP, Pelliccia A, Rollo VC, Chiarelli F, Verrotti A. Panayiotopoulos syndrome: diagnosis and management. Neurol Sci. 2007 Apr;28(2):72-9. doi: 10.1007/s10072-007-0790-4. PMID: 17464469
Graziosi A, Pellegrino N, Di Stefano V, Raucci U, Luchetti A, Parisi P. Misdiagnosis and pitfalls in Panayiotopoulos syndrome. Epilepsy Behav. 2019 Sep;98(Pt A):124-128. doi: 10.1016/j.yebeh.2019.07.016. Epub 2019 Jul 29. PMID: 31369969.
The Tony Stark story
Vous voyez en consultation un homme de 36 ans. Il présente comme antécédents des convulsions hyperthermiques dans l’enfance, une sténose du pylore à 3 semaines de vie, une déficience intellectuelle avec retard développemental. Il habite dans un foyer de vie depuis l’adolescence. Le caryotype et l’IRM cérébrale réalisés à l’âge de 12 ans sont normaux.
Depuis un an, ce patient présente une perte progressive de la marche avec dégradation cognitivo-comportementale, apathie, hypotension orthostatique et une dyspraxie.
A l’examen, vous retrouvez une rigidité extrapyramidale de l’hémicorps gauche qui prédomine au membre supérieur ainsi que des rétractions tendineuses.
Voici une nouvelle IRM cérébrale réalisée quelques jours avant votre consultation.
Quel est votre diagnostic ?
BPAN (Beta propeller protein-associated neurodegeneration)
Il s’agit d’une BPAN pour Beta Propeller protein-Associated Neurodegeneration. Qui est une cause rare de neurodégénération avec accumulation de fer appartenant à la famille des NBIA (Neurodegeneration with Brain Iron Accumulation).
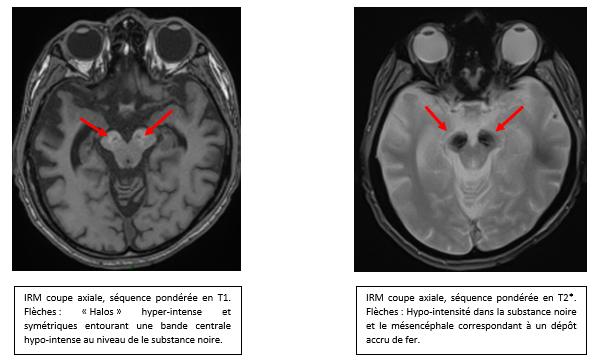
La BPAN est responsable d’un retard psychomoteur, d’une épilepsie, d’une atteinte pyramidale, d’un syndrome extra-pyramidal, d’une déficience intellectuelle et de troubles du comportement de type autistique. Le phénotype peut etre variable, mais l’évolution en deux temps (retard intellectuel dans l’enfance avec épilepsie puis développement de troubles moteurs neurologiques au début de l’âge adulte) doivent évoquer le diagnostic. L’apparition d’image typique à l’IRM cérébrale dans la seconde phase d’évolution, tel que des halos hyper-intenses et symétriques entourant une bande centrale hypo-intense dans la substance noire en séquence pondérée en T1 est pathognomonique des BPAN. On retrouve également sur les séquences T2 et SWI une hypo-intensité dans la substance noire et les pédoncules cérébraux correspondant aux zones de dépots accru de fer.
La transmission est dominante liée à l’X et le diagnostic est établi par l’identification d’une mutation du gène WDR45. Ceci dit, la grande majorité des cas sont des cas sporadiques résultant d’une mutation de novo.
L’IRM cérébrale permet d’évoquer le diagnostic et la mutation
A ce jour il n’y a pas de traitements curatifs des BPAN, mais les chélateurs de fer sont une piste en cours d’exploration.
Bibliographie :
Amaral LLF, Gaddikeri S, Chapman PR, Roy R, Gaddikeri RS, Marussi VH, et al. Neurodegeneration with Brain Iron Accumulation: Clinicoradiological Approach to Diagnosis. J Neuroimaging Off J Am Soc Neuroimaging. août 2015;25(4):539‑51.
Hor CHH, Tang BL. Beta-propeller protein-associated neurodegeneration (BPAN) as a genetically simple model of multifaceted neuropathology resulting from defects in autophagy. Rev Neurosci. 24 avr 2019;30(3):26177.
Un train peut en cacher un autre
Neurologie en raison de mouvements anormaux de nature choréique apparus depuis quelques mois. IL n’y a aucun antécédant familial connu, mais la recherche de mutation du gène IT5 conduit au diagnostic de maladie de Huntington. Un traitement par Tétrabénazine 75 mg/J permet une amélioration significative des mouvements choréiques. Début 2014, le patient est revu pour une consultation de contrôle dans le cadre de sa maladie de Huntington. Il signale des troubles importants de la marche nécessitant depuis quelques mois l’utilisation d’un appui unilatéral.
L’examen neurologique ne note pas de mouvements choréiques. La marche est spastique avec un fauchage bilatéral. IL existe une instabilité bilatérale à la manœuvre de Mingazzini, les réflexes ostéotendineux sont vifs aux membres inférieurs et il existe un signe de Babinski bilatéral.
Une IRM médullaire est réalisée.
- Un bilan complémentaire est- il justifié dans ce contexte ? Si oui lequel ?
- Comment l’interprétez vous ? Que proposez vous ?
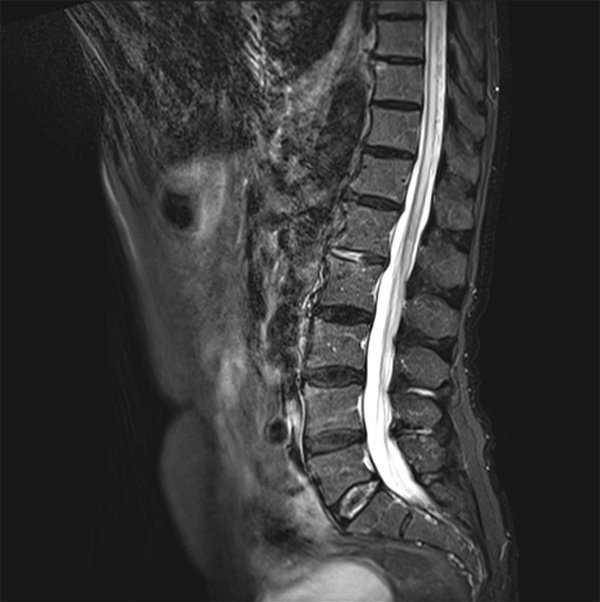
L’IRM médullaire montre un hypersignal intramédullaire étendu de T7 à D1 et des dilatations vasculaires postérieures au niveau du cône terminal en faveur d’une fistule durale.
Une artériographie médullaire est réalisée : l’injection de la 8ème artère intercostale droite opacifie des vaisseaux anormaux au niveau de la moelle épinière correspondant au drainage veineux périmédullaire de la fistule durale.
Une embolisation est programmée.
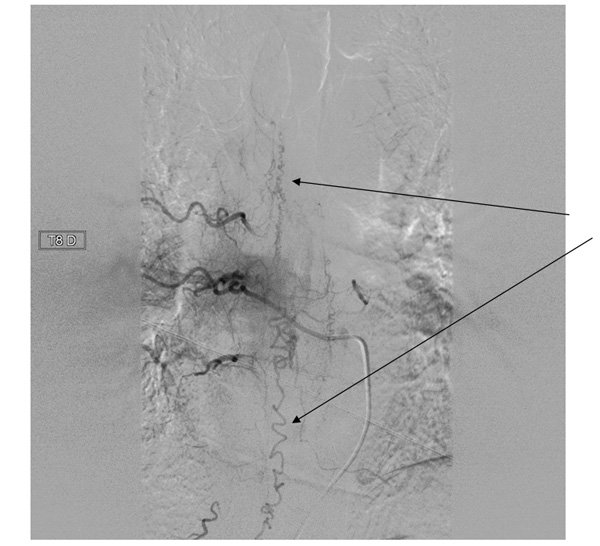
Les fistules durales rachidiennes correspondent à un shunt situé dans la dure mère, entre une artère méningée et le système veineux médullaire . Il s’agit de la plus fréquente malformation vasculaire médullaire. Elle survient préférentiellement chez l’homme entre 50 et 60 ans.
La souffrance médullaire est secondaire à une hypertension veineuse ou à des lésions d’ischémie veineuse. Cette souffrance se manifeste par des signes de claudication médullaire, un syndrome pyramidal, des troubles sphinctériens, des troubles proprioceptifs . L’installation des signes cliniques est souvent insidieuse, mais de rares cas de début brutal (myelopathie ischémique ou rarement hémorragie).
L’IRM médullaire montre le plus souvent au niveau du cône terminal médullaire, un hypersignal T2 médullaire étendu et un élargissement de la moelle (œdème médullaire), et des images serpigineuses correspondant à des veines périmédullaires dilatées (hypointenses en T2 se rehaussant après injection de gadolinium T1). Ces signes n’ont pas de valeur localisatrice de la fistule (qui peut être cervicale, thoracique, lombaire).
Une artériographie médullaire met en évidence la fistule dont le traitement est endovasculaire (embolisation)
Pour en savoir plus :
Hacein-Bey L, Konstas AA, Pile-Spellman J Natural history, current concepts, classification, factors impacting endovasculartherapy, and pathophysiology of cerebral and spinal dural arteriovenous fistulas Clin Neurol Neurosurg. 2014 Jun;121:64-75.
Jellema K, Tijssen CC, van Gijn J. Spinal dural arteriovenous fistulas: a congestive myelopathy that initially mimics a peripheral nerve disorder Brain. 2006 Dec;129(Pt 12):3150-64.
Yen PP, Ritchie KC, Shankar JJ. Spinal dural arteriovenous fistula: correlation between radiological and clinical findings. J Neurosurg Spine. 2014 Aug 15:1-6. [Epub ahead of print]
Une explosion dans la tête
C’est un homme de 30 ans qui consulte pour « un sentiment de déflagration dans la tête ».
Cette symptomatologie évolue depuis l’âge de 16-17 ans, à raison de 3 à 4 épisodes par an. C’est le soir, au moment de l’endormissement que surviennent ces épisodes qu’il décrit en regard du vertex, paracentral droit, discrètement en arrière, ne prenant pas toute la tête. Il les ressent comme « choquant, très bizarre et très surprenant». L’accès est bref, non associé à une gêne ou une douleur par la suite. Le réendormissement est plutôt facile.
Quel est votre diagnostic ?
- Syndrome de la tête qui explose
- Variante explosive du ice pick headache
- Céphalée hypnique
- Forme atypique de myoclonies d’endormissement
- Crise focale hypnique
Le syndrome de la tête qui explose ; exploding head syndrome
Le syndrome de la tête qui explose figure dans le chapitre « autres parasomnies » dans la classification internationale des troubles du sommeil (ICSD-III).
Il s’agit de la perception d’un bruit soudain ou l’impression d’une violente explosion dans la tête à l’endormissement ou lors d’un réveil nocturne. L’événement entraîne un réveil brutal souvent associé à un sentiment de peur. L’expérience n’est pas associée à des plaintes importantes de douleur et ne nécessite pas de traitement particulier.
Les mécanismes neurophysiologiques sous-jacents sont inconnus.
L’évolution est bénigne. En cas d’événements répétés dans la même nuit, le syndrome de la tête qui explose peut entraîner une insomnie. Le traitement repose alors sur une bonne hygiène du sommeil et une approche cognitivo-comportemental de l’insomnie.
American Academy of Sleep Medicine (AASM). The International classification of sleep disorders: diagnostic & coding manual (ICSD-3). 3rd edn. Darien, IL: American Academy of Sleep Medicine; 2014.
Exploding head syndrome: clinical features, theories about etiology, and prevention strategies in a large international sample. Sharpless BA, Denis D, Perach R, French CC, Gregory AM. Sleep Med. 2020 Nov;75:251-255. doi: 10.1016/j.sleep.2020.05.043. Epub 2020 Jun 10. PMID: 32862013
Exploding Head Syndrome: a Review. Ceriani CEJ, Nahas SJ. Curr Pain Headache Rep. 2018 Jul 30;22(10):63. doi: 10.1007/s11916-018-0717-1. PMID: 30062616
15/06/2023
admin
15/06/2023
admin
15/06/2023
admin
15/06/2023
admin
Références
Keddie S, Adams A, Kelso ARC, Turner B, Schmierer K, Gnanapavan S, Malaspina A, Giovannoni G, Basnett I, Noyce AJ. No laughing matter: subacute degeneration of the spinal cord due to nitrous oxide inhalation. J Neurol. 2018 May;265(5):1089-1095. doi: 10.1007/s00415-018-8801-3. Epub 2018 Mar 3. PMID: 29502317; PMCID: PMC5937900.
Smith SE, Kinney HC, Swoboda KJ, Levy HL. Subacute combined degeneration of the spinal cord in cblC disorder despite treatment with B12. Mol Genet Metab. 2006 Jun;88(2):138-45. doi: 10.1016/j.ymgme.2006.02.007. Epub 2006 Mar 30. PMID: 16574454.
Noh T, Osman G, Chedid M, Hefzy H. Nitrous oxide-induced demyelination: Clinical presentation, diagnosis and treatment recommendations. J Neurol Sci. 2020 Jul 15;414:116817. doi: 10.1016/j.jns.2020.116817. Epub 2020 Apr 3. PMID: 32302804.