Situations de départ
 12 Nausées.
12 Nausées.
13 Vomissements.
28 Coma et troubles de la conscience.
50 Malaise et perte de connaissance.
64 Vertiges et sensation vertigineuse.
66 Difficulté à la marche.
74 Faiblesse musculaire.
118 Céphalées.
119 Confusion mentale/désorientation.
120 Convulsions.
121 Déficit neurologique sensitif et/ou moteur.
127 Paralysie faciale.
134 Troubles du langage et/ou phonation.
138 Anomalie de la vision.
226 Découverte d’une anomalie du cerveau à l’examen d’imagerie médicale.
230 Rédaction de la demande d’un examen d’imagerie.
Objectifs pédagogiques
Diagnostiquer une tumeur intracrânienne.
Identifier les situations d’urgence et planifier leur prise en charge.
Hiérarchisation des connaissances
| Rang | Rubrique | Intitulé | Descriptif |
|---|---|---|---|
 |
Définition | Principaux types et localisations des tumeurs intracrâniennes | Savoir distinguer : tumeur primitive/secondaire, de l’encéphale/des annexes, bénigne/maligne, fréquente/rare, sus- ou sous-tentorielle |
|
Définition | Tumeurs primitives intracrâniennes : système nerveux central (SNC) et annexes | Connaître la distinction entre tumeur provenant du SNC et tumeur provenant de ses annexes |
 |
Définition | Connaître les principaux types histologiques des tumeurs cérébrales | Méningiome et adénome hypophysaire/ tumeurs gliales de bas grade et de haut grade (glioblastome)/métastases |
|
Prévalence, épidémiologie | Tumeurs primitives Intracrâniennes : différents types | Connaître les principaux types de tumeurs primitives intracrâniennes et leur origine |
|
Prévalence, épidémiologie | Tumeurs secondaires intracrâniennes : métastases cérébrales | Décrire la prévalence relative des métastases cérébrales, leur apparition dans l’histoire clinique du cancer et les principaux sites primitifs pourvoyeur de métastases cérébrales |
|
Diagnostic positif (examen clinique, démarche diagnostique) | Formes et symptômes cliniques | Connaître les principaux tableaux cliniques devant faire évoquer une tumeur intracrânienne |
|
Examens complémentaires | Connaître la stratégie d’exploration en imagerie devant une tumeur intracrânienne de l’adulte | Le scanner et l’IRM sans et avec injection permettent d’évoquer le diagnostic de tumeur, mais l’IRM est plus performante pour le diagnostic et le bilan préopératoire |
|
Diagnostic positif (examen clinique, démarche diagnostique) | Tumeurs secondaires intracrâniennes : recherche du cancer primitif | Décrire la recherche systématique de cancer primitif à effectuer devant une métastase cérébrale |
|
Identifier une urgence | Savoir évoquer une hypertension intracrânienne (HTIC), une épilepsie chez un patient porteur d’une tumeur intracérébrale | Connaître les deux principales urgences (HTIC, épilepsie) révélant ou compliquant l’évolution d’une tumeur cérébrale |
|
Suivi et/ou pronostic | Connaître les principes de la prise en charge de l’HTIC et de l’épilepsie chez un patient porteur d’une tumeur intracérébrale |
475 La prise en charge des patients atteints de tumeurs intracrâniennes implique un partenariat multidisciplinaire où neuro-oncologue, neurochirurgien, neuroradiologue, anatomopatho-logiste, radiothérapeute, médecin généraliste, médecin de soins palliatifs ont leur place.
I Épidémiologie des tumeurs intracrâniennes de l’adulte
A Tumeurs primitives
- • L’incidence des tumeurs intracrâniennes primitives de l’adulte est de 10 cas pour 100 000 habitants par an (6 000 nouveaux cas par an) en France.
- • Par ordre décroissant, il s’agit de méningiomes (40 % des tumeurs cérébrales primitives), de gliomes tous grades confondus (30 %) et d’adénomes hypophysaires (10 %).
- • Les tumeurs cérébrales malignes représentent un tiers d’entre elles, soit 1 % de l’ensemble des cancers.
B Métastases
- • Les métastases cérébrales constituent la grande majorité des tumeurs intracrâniennes, tous types confondus.
- • 476Leur incidence est sous-évaluée, car ces tumeurs restent souvent asymptomatiques du vivant des patients.
- • Les études autopsiques suggèrent qu’elles compliquent l’évolution de 20 % des cancers.
- • En dehors des cancers du poumon, où les métastases cérébrales peuvent être révélatrices du cancer dans 20 % des cas, celles-ci surviennent tardivement dans l’histoire naturelle du cancer, et trois quarts des patients ont déjà des métastases dans d’autres localisations.
II Neuropathologie
A Tumeurs primitives
Le diagnostic de certitude repose sur l’analyse histologique d’un fragment tumoral obtenu par biopsie chirurgicale ou lors d’un geste d’exérèse.
La classification utilisée est celle de l’OMS qui distingue les tumeurs en fonction de leur cellule d’origine (astrocyte, oligodendrocyte…), de leur grade de malignité (nombre de cellules en mitoses, prolifération endothélocapillaire et foyers de nécrose) et la présence d’anomalies chromosomiques ou moléculaires dans les cellules tumorales (tableau 26.1).
Classification histopronostique de l’OMS (2021) des tumeurs cérébrales primitives.
| Type tumoral | Sous-types |
|---|---|
| Gliomes diffus | Oligodendrogliome de grade II et III (codélétion 1p/19q) Astrocytome diffus avec IDH muté Grade II, III et IV Glioblastome (grade IV) Gliome diffus de la ligne médiane (mutation histone) |
| Tumeurs épendymaires | Subépendymome, épendymome |
| Tumeurs gliales, neuronales et neurogliales circonscrites | Astrocytome pilocytique, gangliogliome, tumeur neuroépithéliale dysembryoplasique (DNET), neurocytome central et autres (grade I) |
| Tumeurs de la région pinéale | Pinéalocytome, pinéaloblastome, tumeur du parenchyme pinéal, tumeur papillaire de la région pinéale |
| Tumeurs du plexus choroïde | Papillome, carcinome du plexus choroïde |
| Tumeurs embryonnaires | Médulloblastome |
| Tumeurs germinales | Germinomes, carcinome embryonnaire, choriocarcinome, tératome… |
| Méningiomes et tumeurs mésenchymateuses non méningothéliales | Tumeur fibreuse solitaire, hémangioblastome, sarcome, et autres |
| Tumeurs de la région sellaire | Adénome hypophysaire Craniopharyngiome |
| Lymphome primitif du système nerveux central | |
DNET : Dysembryoplastic NeuroEpithelial Tumors.
Source : Laurenge A, Tran S, Peyre M, Idbaih A. Classification OMS 2021 des tumeurs du système nerveux central. Traité Neurologie, 01 Jul 2023, Vol. 46, Issue 3, pages 1-17. DOI : 10.1016/S0246-0378(23)68992-4
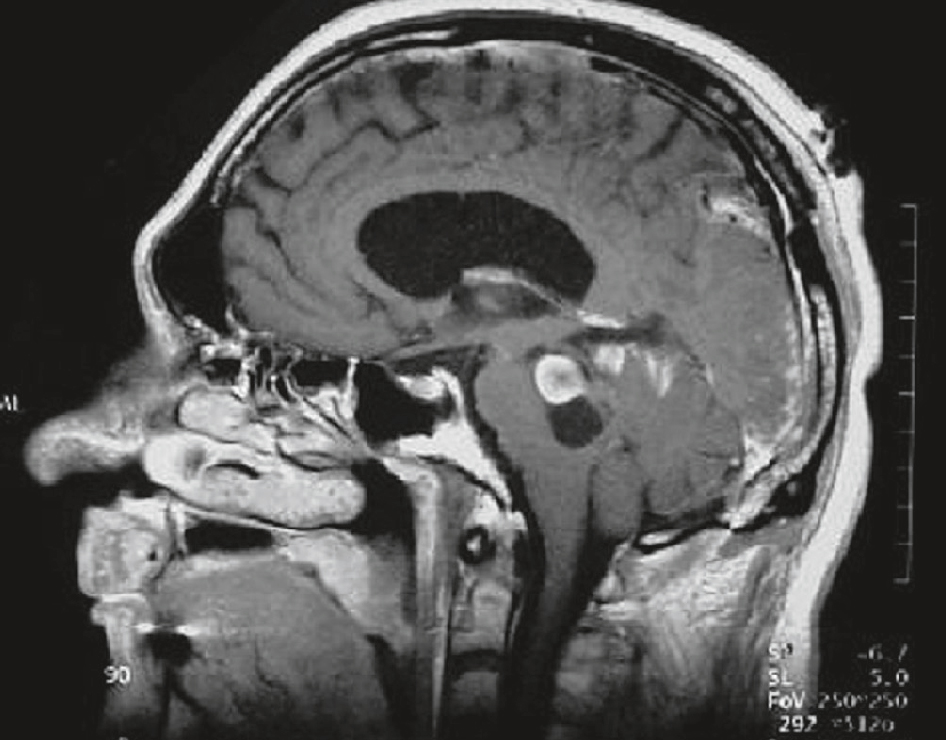
IRM cérébrale d’un patient de 22 ans présentant un tableau d’HTIC. Séquence T1 avec injection de gadolinium, coupe sagittale : lésion du tronc cérébral constituée d’une partie nodulaire prenant le contraste et d’une portion kystique (hyposignal). Hydrocéphalie (dilatation ventriculaire).
Image d’IRM cérébrale en coupe sagittale chez un patient de 22 ans présentant un tableau d’hypertension intracrânienne (HTIC). On observe une volumineuse lésion arrondie, hyperintense et bien circonscrite, située dans la région du tronc cérébral, probablement au niveau du mésencéphale ou du pont, avec un effet de masse visible sur les structures adjacentes. Cette masse entraîne un écrasement des espaces sous-arachnoïdiens et une déformation des ventricules, traduisant une obstruction possible à l’écoulement du liquide cérébrospinal, compatible avec une HTIC. Le parenchyme cérébral environnant semble refoulé, ce qui souligne la pression exercée par la lésion intracrânienne.
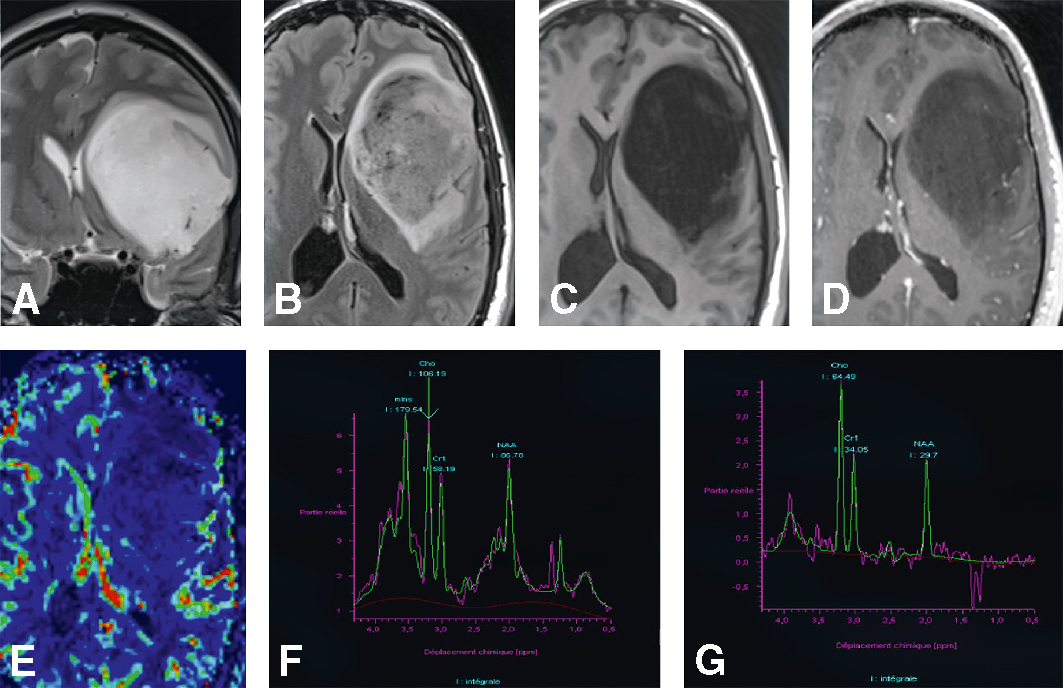
IRM cérébrale chez un patient de 26 ans présentant des crises épileptiques partielles depuis plusieurs mois. Volumineuse lésion paralimbique gauche en hypersignal T2 (A), en hypersignal hétérogène FLAIR (B), en hyposignal T1 (C), sans rehaussement pathologique (D), sans signes de néoangiogenèse : rCBV à 0,5 (E), avec un spectre tumoral : rapport choline/créatine (Cho/Cr) et choline/N-acétyl-aspartate (Cho/NAA) augmenté, présence de myo-inositol (ml) (F, G). A. T2, coupe coronale. B. FLAIR, coupe axiale. C. T1, coupe axiale. D. T1 avec injection de gadolinium, coupe axiale. E. Séquence de perfusion. F, G. Spectroscopie par résonance magnétique monovoxel à écho court 30 ms (F) et à écho long 135 ms (G).
Cette série d’IRM cérébrales concerne un patient de 26 ans souffrant de crises épileptiques partielles chroniques. L’image A en T2 montre une masse volumineuse hyperintense dans la région fronto-pariétale droite, exerçant un effet de masse sur les structures médianes. Les images B à D en FLAIR, T1 avant et après injection de gadolinium révèlent une lésion hétérogène avec rehaussement périphérique, évoquant un processus tumoral infiltrant. L’image E, en cartographie de perfusion, met en évidence une augmentation focale du débit sanguin suggérant une néoangiogenèse tumorale. Les spectroscopies F et G révèlent des profils métaboliques avec élévation de la choline et diminution du NAA, renforçant l’hypothèse d’une tumeur gliale de haut grade.
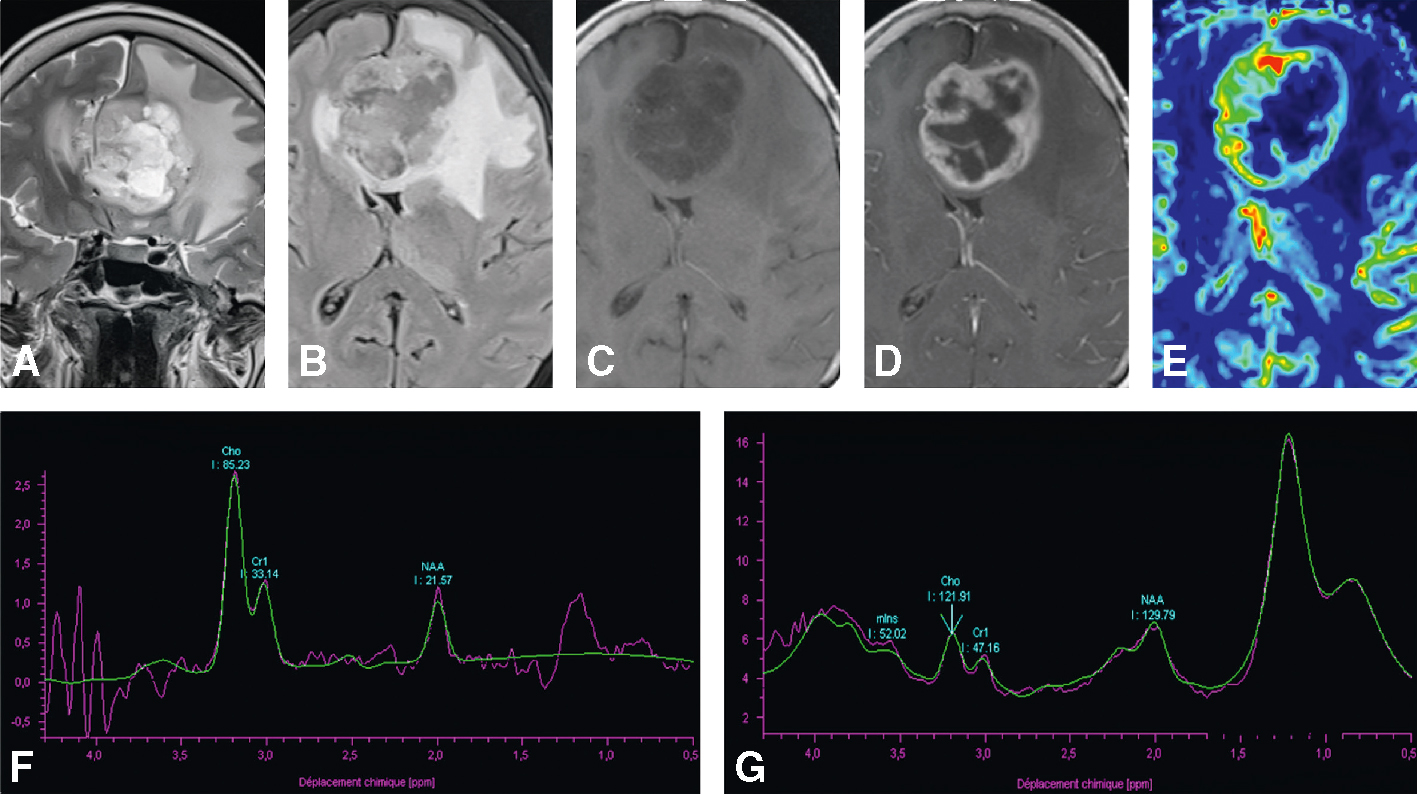
IRM cérébrale chez une patiente de 67 ans, présentant des céphalées et des troubles phasiques d’installation rapide. Lésion bi-fronto-calleuse en hypersignal hétérogène T2 (A), en signal hétérogène FLAIR et œdème périlésion-nel important (B), en hyposignal T1 (C), avec rehaussement périphérique hétérogène et centre nécrotique (D), des signes francs de néoangiogenèse : rCBV à 3,6 (E), et un spectre tumoral : rapport choline/créatine (Cho/ Cr) et choline/N-acétyl-aspartate (Cho/NAA) augmenté, présence de lipides et lactates (F, G). A. T2, coupe coronale. B. FLAIR, coupe axiale. C. T1, coupe axiale. D. T1 avec injection de gadolinium, coupe axiale. E. Séquence de perfusion. F, G. Spectroscopie par résonance magnétique monovoxel à écho court 30 ms (F) et à écho long 135 ms (G).
IRM cérébrale d’une patiente de 67 ans souffrant de céphalées et de troubles phasiques brusques, révélant une masse intra-parenchymateuse. L’image A montre une lésion hétérogène en hypersignal T2 avec effet de masse important. L’image B, en T1 sans contraste, présente une hypointensité centrale, tandis que l’image C, en séquence FLAIR, révèle un hypersignal périlésionnel traduisant un œdème. L’image D, après injection de gadolinium, met en évidence un rehaussement annulaire épais et irrégulier. L’image E, une cartographie en perfusion, montre une hyperperfusion périphérique. Les spectroscopies F et G indiquent un pic de choline élevé, une diminution du NAA et un pic de lactate, suggérant un processus tumoral agressif de type glioblastome.
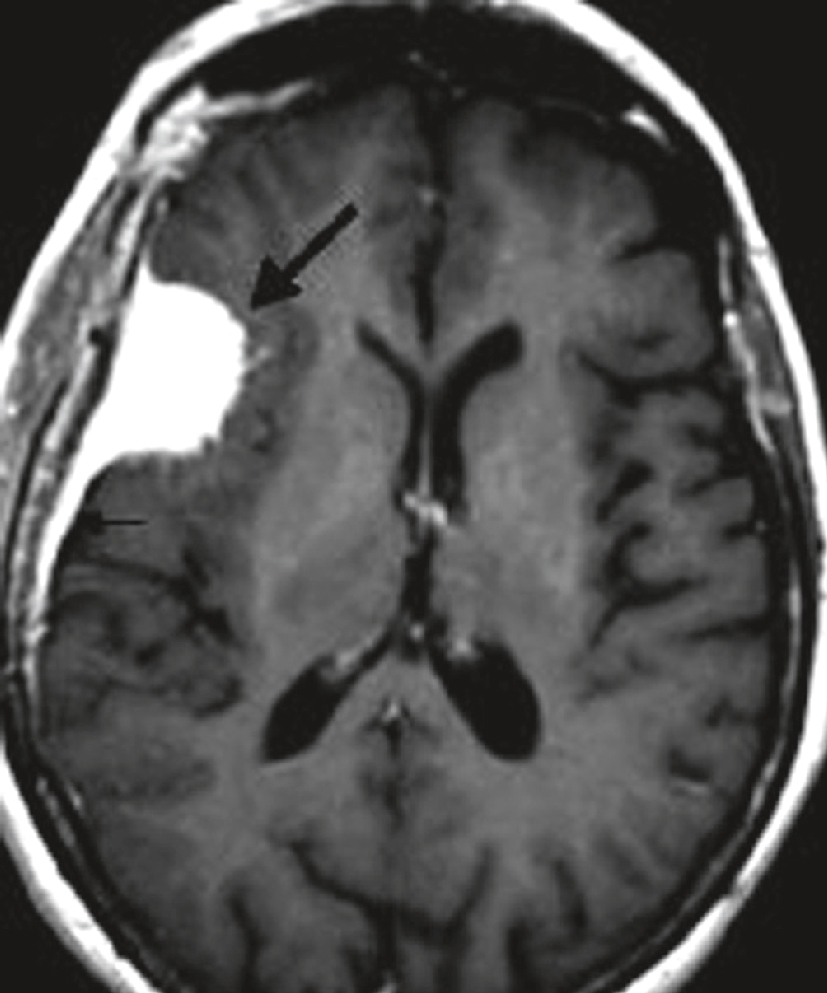
IRM cérébrale chez une femme de 54 ans présentant des crises d’épilepsie partielle depuis 1 mois. Séquence axiale T1 après injection de gadolinium : lésion extracérébrale, refoulant le parenchyme cérébral, bien limitée, à base d’implantation large sur la convexité, prenant le contraste de manière intense et homogène (flèche en gras). Prise de contraste de la dure-mère adjacente (petite flèche) correspondant à la languette d’insertion dure-mérienne du méningiome (image en « queue de comète »).
IRM cérébrale en coupe axiale pondérée en T1 montrant une masse cortico-sous-corticale hyperintense bien délimitée au niveau du lobe frontal gauche, avec un effet de masse sur le ventricule latéral adjacent et une discrète hypodensité périlésionnelle compatible avec un œdème vasogénique. La lésion, indiquée par une flèche épaisse, évoque une tumeur d’allure infiltrante responsable de la déformation des structures avoisinantes. La flèche fine signale un effet de masse modéré sur le cortex adjacent. Le contexte clinique de crises d’épilepsie partielle récentes chez cette patiente de 54 ans renforce la suspicion d’un processus tumoral évolutif, tel qu’un gliome de bas ou de haut grade.
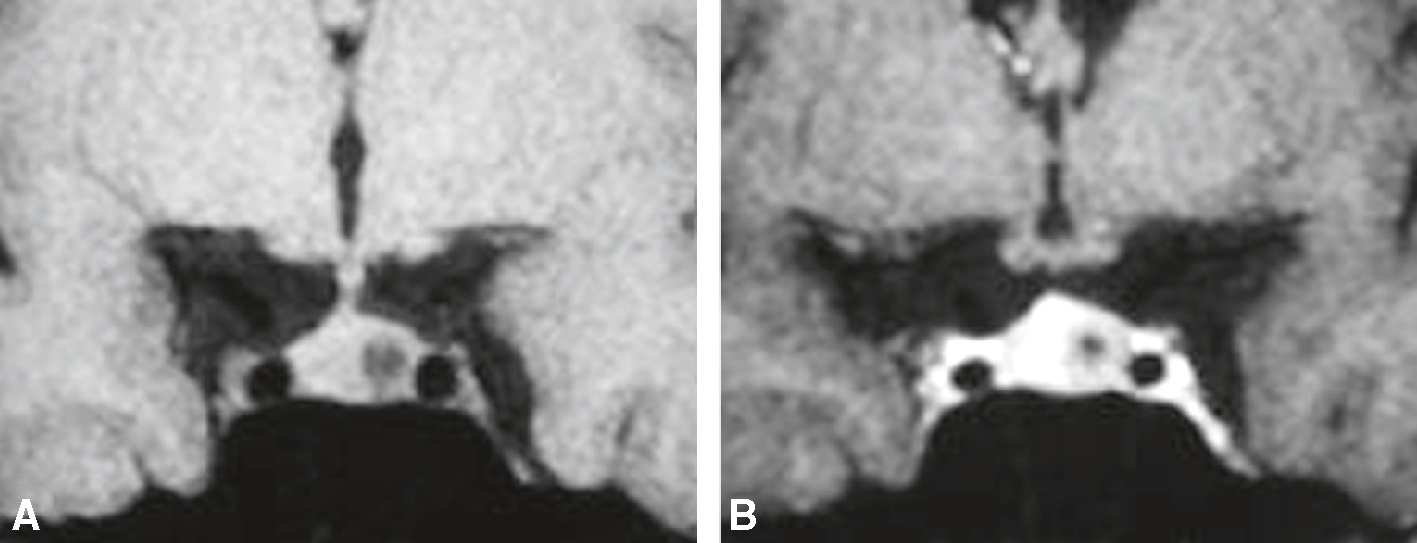
IRM, séquence T1 dans le plan coronal, avant et après injection de gadolinium chez une femme de 45 ans réalisée dans le cadre d’un bilan d’hyperprolactinémie. Signes directs : lésion intrasellaire focale arrondie, infracentimétrique, développée dans l’aileron hypophysaire gauche, en hyposignal avant injection et restant en hyposignal après injection (rehaussement beaucoup moins rapide que l’hypophyse normale). Signes indirects : surélévation modérée du diaphragme sellaire à gauche et déviation controlatérale de la tige pituitaire. A. T1 frontal sans injection. B. T1 frontal après injection.
Deux trophées IRM coronaux dans la séquence T1 montrent la zone de la glande hypophysaire d'un patient de 45 ans examiné pour l'hyperprolactinémie. Avant d'injecter du gadolinium, cette photographie mettait en vedette la glande hypophysaire généralement iso-centrique avec une apparence asymétrique séparée de la boucle avant. L'image B recevant l'injection après l'injection met en évidence le contraste hétérogène dans la glande pituitaire avec des zones de réadaptation du lobe bas de l'hypophyse, indiquant la présence de la glande pituitaire microadénomes. Cette amélioration différentielle permet une meilleure distinction des lésions du parenchyme hypophysaire normal. Cela conduit au diagnostic de l'adénome de la prolactine, une cause commune d'hyperprolactinémie pendant les services de garde.
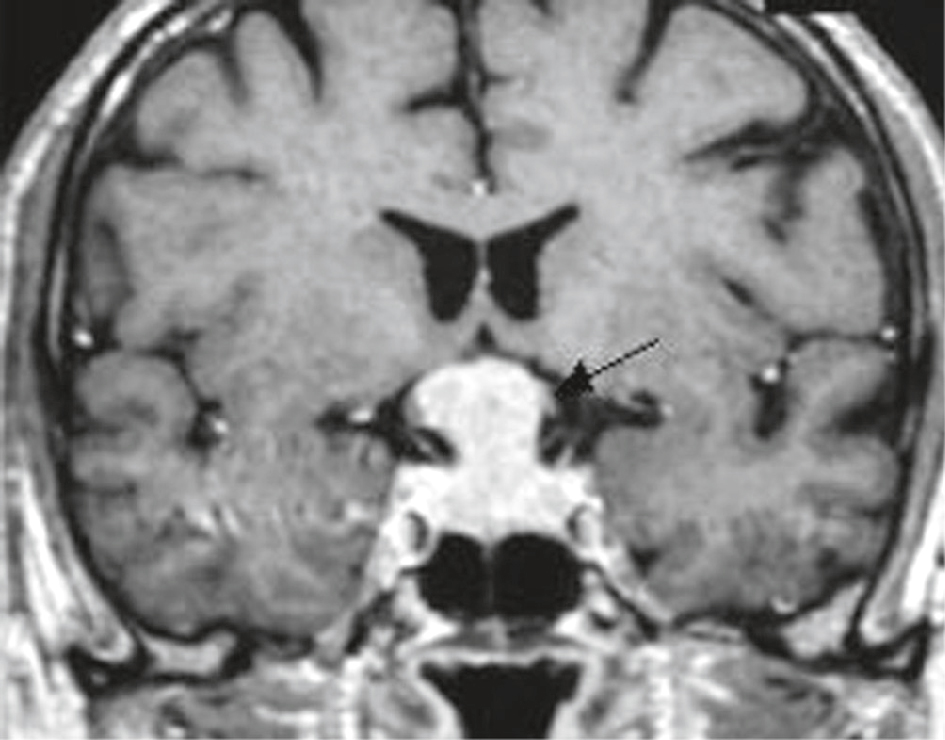
IRM cérébrale chez une femme de 57 ans avec hémianopsie bitemporale. Séquence T1 après injection dans le plan coronal : lésion à développement intrasellaire et à extension supra-sellaire, en forme de « bonhomme de neige » ou de « 8 » (forme liée à la constriction du diaphragme sellaire), prenant le contraste de manière homogène. La tige pituitaire n’est plus visible. Important refoulement du chiasma optique (flèche). Pas d’envahissement des loges caverneuses.
Image en coupe coronale d’une IRM cérébrale montrant une patiente de 57 ans présentant une hémianopsie bitemporale, dans un contexte évocateur d’une atteinte du chiasma optique. On observe une masse isointense en hypersignal T1 située juste au-dessus de la selle turcique, compriment la région chiasmatique. La flèche indique précisément la zone de compression à l’origine probable du déficit visuel bilatéral temporal. La morphologie des ventricules latéraux reste conservée, sans signe d’hydrocéphalie associée. L’ensemble de l’architecture cérébrale reste globalement symétrique, en dehors de cette lésion sellarienne typiquement compatible avec un adénome hypophysaire comprimant le chiasma optique.
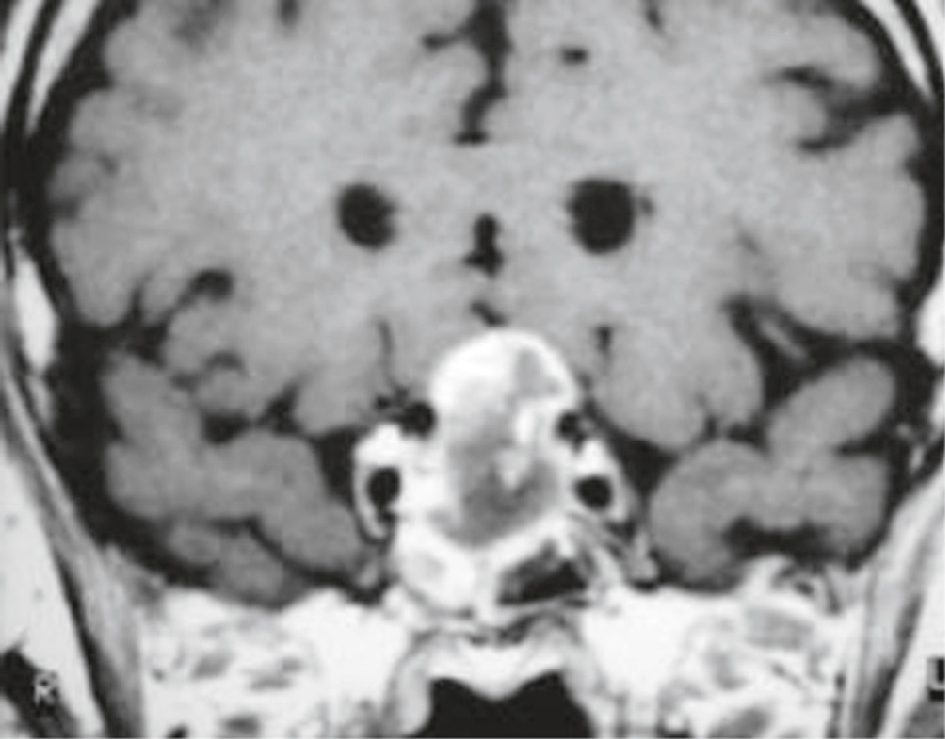
IRM cérébrale chez un homme de 72 ans avec céphalées et hémianopsie bitemporale. Séquence frontale T1 après injection : macro-adénome invasif, à développement intra-, supra- et infrasellaire. Envahissement de la loge caverneuse droite et du sinus sphénoïdal. Prise de contraste hétérogène, avec remaniements nécrotiques centraux (zones en hyposignal).
Image en coupes coronales d’une IRM cérébrale pondérée en T1 réalisée chez un patient de 72 ans présentant des céphalées et une hémianopsie bitemporale. La séquence met en évidence une masse isointense située au niveau de la région suprasellaire, comprimant les structures chiasmatiques. Le chiasma optique apparaît refoulé vers le haut, ce qui explique le déficit visuel bilatéral dans les champs temporaux. Le contour tumoral est net, suggérant une lésion expansive bien délimitée, probablement un adénome hypophysaire. Le parenchyme cérébral adjacent montre un aspect normal, sans signes d’œdème ni d’effet de masse notable.
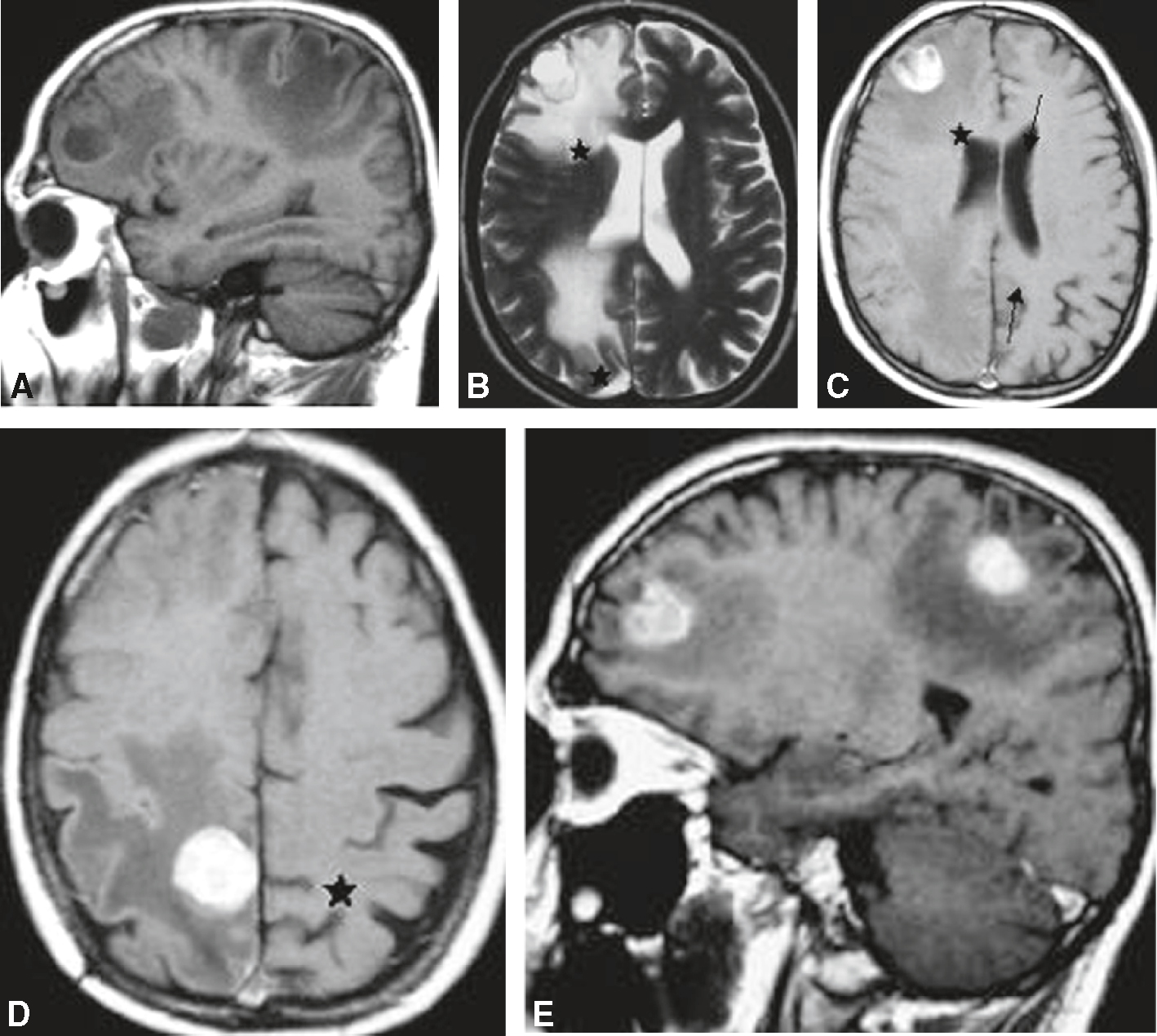
IRM cérébrale réalisée chez un patient de 67 ans traité pour un adénocarcinome bronchique avec une hémiparésie gauche récente. Présence de deux lésions intracérébrales hémisphériques droites, bien limitées et arrondies, développées respectivement dans la région frontale antérieure et au niveau du lobule paracentral (région centrale interne), en hyposignal T1 et en hypersignal T2, entourées d’un œdème important. Effet de masse sur le ventricule latéral droit avec effacement partiel des cornes frontale et occipitale. Après injection, important rehaussement des deux lésions permettant de les distinguer de l’œdème. A. T1 sagittal. B. T1 axial. C. T1 axial après injection. D. T1 axial après injection. E. T1 sagittal après injection.
Série d’IRM cérébrales réalisées chez un patient de 67 ans atteint d’un adénocarcinome bronchique, présentant une hémiparésie gauche récente. L’image A en coupe sagittale montre une masse ronde bien délimitée en région frontale droite avec œdème périlésionnel, associée à une discrète prise de contraste. Les coupes axiales B et C révèlent plusieurs métastases, identifiables par leur hypersignal en T2 avec effet de masse et déplacement des structures médianes. La présence d’un important œdème vasogénique autour des lésions est visible, contribuant à la symptomatologie. Les images D et E confirment l’aspect nodulaire des métastases avec rehaussement périphérique après injection de gadolinium, suggérant une atteinte secondaire du parenchyme cérébral d’origine néoplasique.
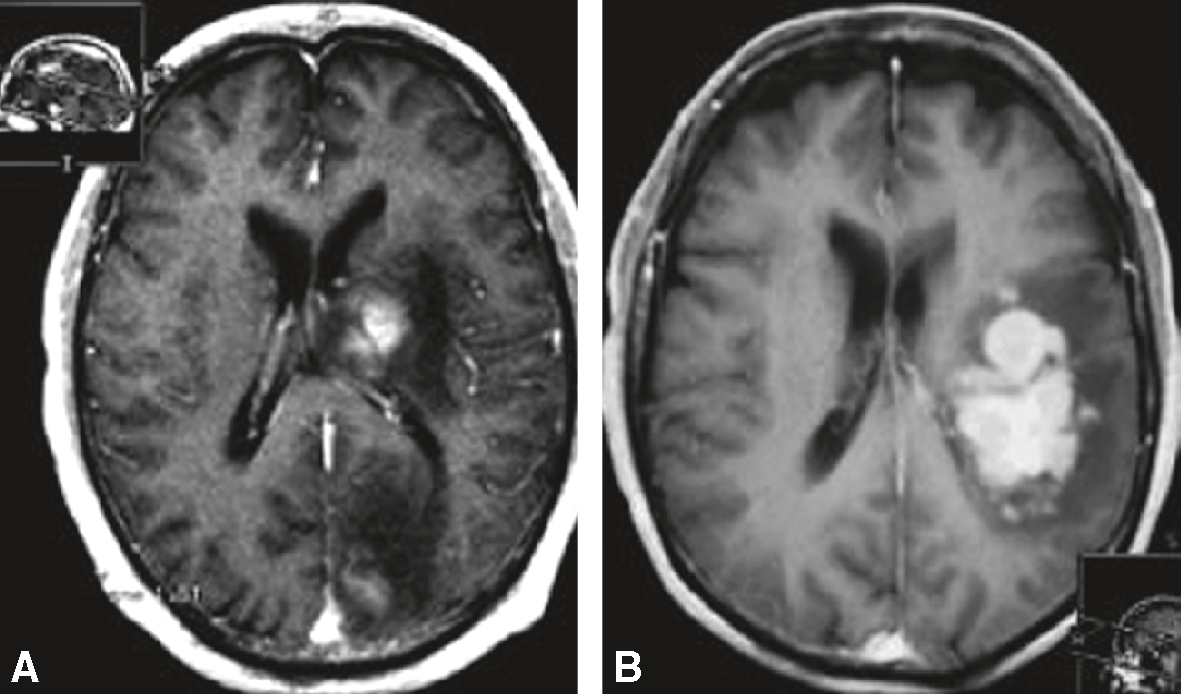
Lésions intracérébrales généralement paraventriculaires ou près des espaces sous-arachnoïdiens, prenant fortement le contraste de manière homogène. Les lymphomes sont très sensibles à la corticothérapie et les images peuvent « disparaître » en délai bref (1 ou 2 semaines), rendant difficile le repérage stéréotaxique, mais aussi l’interprétation anatomopatholo-gique. A. Exemple 1. IRM cérébrale chez une patiente de 71 ans avec ralentissement idéomoteur depuis quelques mois; séquence axiale T1 après injection. B. Exemple 2. IRM cérébrale chez un patient présentant une confusion récente; séquence axiale T1 après injection.
Les deux coupes axiales T1 injectées (A et B) montrent des lésions nodulaires intracérébrales, situées de manière prédominante dans la région paraventriculaire droite, en contact avec le corps calleux et les structures périventriculaires. Sur l’image A, la lésion apparaît en iso- à léger hypersignal avec un effet de masse modéré. Sur l’image B, après injection de gadolinium, on observe une prise de contraste intense, homogène et bien délimitée. Ce type d’aspect est évocateur de lésions d’origine infectieuse ou tumorale, souvent observées dans des contextes d’immunodépression, comme les lymphomes cérébraux primitifs ou les toxoplasmoses cérébrales.
B Métastases
En pratique, le diagnostic de métastases cérébrales peut être retenu sur les caractéristiques de l’IRM cérébrale et ne nécessite pas obligatoirement de confirmation histologique si le cancer 477systémique est connu, en particulier s’il est évolutif et s’il existe d’autres localisations métasta-tiques. Dans le cas contraire, un examen histologique est nécessaire.
Les cancers du poumon et du sein sont à l’origine de plus de la moitié des métastases cérébrales, loin devant les cancers du rein, de l’appareil digestif et des mélanomes. Une IRM cérébrale doit systématiquement être réalisée au cours du bilan d’extension initial des cancers bronchiques à petites cellules et est recommandée chez tous les patients avec cancers bronchiques non à petites cellules.
III Présentation clinique
A Syndrome d’hypertension intracrânienne
Lié à la croissance du processus expansif intracrânien et de l’œdème périlésionnel ou consécutif à un blocage des voies d’écoulement du liquide cérébrospinal (LCS) responsable d’une hydrocéphalie, le syndrome d’hypertension intracrânienne (HTIC) est caractérisé par l’association de :
- • céphalées typiquement matinales;
- • vomissements;
- • œdème papillaire au fond d’œil;
- • une diplopie par atteinte du nerf abducens, qui peut parfois survenir, sans valeur localisatrice.
Chez le nourrisson, le syndrome d’HTIC comporte plusieurs signes spécifiques :
- • une macrocrânie : toujours présente dans les hydrocéphalies chroniques se révélant avant l’âge de 2 ans, elle est caractérisée par un périmètre crânien > 2 DS (déviations standard) au-dessus de la normale; souvent révélatrice, elle peut être remarquée à l’occasion d’une rupture de la courbe de croissance du périmètre crânien;
- • la tension marquée de la fontanelle antérieure et la disjonction des sutures, perceptibles à l’examen;
- • le regard « en coucher de soleil » correspondant à une déviation permanente vers le bas des globes oculaires; la paupière supérieure est rétractée. Une baisse d’acuité visuelle pouvant conduire à la cécité par atrophie optique peut être une conséquence dramatique de l’hydrocéphalie méconnue.
B Crises d’épilepsie
Une crise épileptique partielle ou généralisée est révélatrice de 20 à 40 % des tumeurs cérébrales.
L’épilepsie est plus fréquente pour les tumeurs corticales d’évolution lente (gliome de bas grade, méningiome).
C Déficits focaux
- • Liés à la compression ou à l’infiltration du parenchyme cérébral par la tumeur.
- • Le déficit est aggravé en cas d’œdème vasogénique tumoral associé.
- • Son mode d’apparition est généralement rapidement progressif, s’étendant « en tache d’huile ». Son type dépend de la topographie tumorale.
D 478Troubles cognitifs
Syndrome confusionnel ou démentiel progressif sur quelques semaines en cas de lésions multiples (métastases) ou étendues (gliome infiltrant ou lymphome cérébral).
E Troubles de l’équilibre et atteinte des nerfs crâniens
- • Ataxie cérébelleuse : tumeur du cervelet.
- • Atteinte multiple des nerfs crâniens : tumeur du tronc cérébral.
IV Complications évolutives constituant des urgences thérapeutiques
A Hémorragie intratumorale
Elle est plus fréquente avec certains types de tumeurs (métastases de mélanome ou de cancer du rein) et peut être prise pour un hématome cérébral spontané quand la tumeur sous-jacente n’est pas connue. Elle se manifeste par une HTIC, un trouble de la conscience ou un déficit neurologique focal aigus. Il faut donc demander une IRM cérébrale à 4 à 6 semaines devant une hémorragie parenchymateuse spontanée pour rechercher une tumeur sous-jacente masquée par les signes aigus de l’hémorragie.
B 479Hydrocéphalie
Elle peut résulter :
- • de l’obstruction des voies d’écoulement du LCS par le processus tumoral; l’hydrocéphalie est alors non communicante et toute ponction lombaire est contre-indiquée à cause du risque d’engagement occipital;
- • d’une dissémination tumorale leptoméningée entravant la résorption du LCS; l’hydrocéphalie est alors communicante et autorise une éventuelle ponction lombaire;
- • d’une hypersécrétion du LCS, qui peut être observée dans les tumeurs du plexus choroïde.
Elle peut affecter :
- • une partie du système ventriculaire : par exemple, hydrocéphalie triventriculaire par obstruction de l’aqueduc de Sylvius (aqueduc du mésencéphale);
- • ou l’ensemble de celui-ci : hydrocéphalie tétraventriculaire, secondaire par exemple à une obstruction des trous de Magendie et de Luschka (ouvertures médiane et latérales) du quatrième ventricule, ou à un obstacle à la résorption du LCS lié à une méningite tumorale.
C Engagement
L’engagement cérébral correspond au passage d’une partie du parenchyme cérébral à travers une structure rigide de l’encéphale (tente du cervelet, trou occipital, etc.).
1 Engagement temporal
Il correspond au passage des structures temporomésiales dans la fente de Bichat (fissure transverse du cerveau), entre le bord libre de la tente du cervelet et le tronc cérébral. Il doit être suspecté devant l’apparition d’une paralysie (souvent partielle) du nerf oculomoteur (III) homolatéral avec ptosis, mydriase aréactive, s’accompagnant d’une hémiparésie controlatérale à la tumeur.
2 Engagement des amygdales cérébelleuses dans le trou occipital
C’est une complication gravissime des processus sous-tentoriels. Un port guindé de la tête, un torticolis doivent faire craindre sa survenue. Son risque est la compression de la moelle allongée (bulbe) qui entraîne une déficience respiratoire majeure avec des mouvements de décérébration (enroulement des membres supérieurs et opisthotonos) ou une mort subite.
D Méningite tumorale
Elle résulte de l’extension aux espaces sous-arachnoïdiens d’une tumeur cérébrale primitive ou de l’invasion par métastases d’un cancer systémique. Cliniquement, le diagnostic est suspecté devant une paralysie d’un ou plusieurs nerfs crâniens d’installation rapidement progressive, des douleurs rachidiennes souvent associées à des douleurs radiculaires et/ou une aréflexie, des troubles de l’équilibre, des céphalées, une atteinte des fonctions cognitives souvent fluctuante. C’est surtout la combinaison de ces signes qui est évocatrice, témoignant d’un processus multifocal. En revanche, la raideur méningée est plus rare. Le diagnostic repose sur :
- • l’IRM cérébrale et l’IRM médullaire, qui peuvent mettre en évidence des prises de contraste méningées ou périventriculaires anormales très évocatrices, mais aussi dans la région thoracolombaire;
- • la ponction lombaire (en l’absence de contre-indication) à la recherche de cellules tumorales (qu’il faut analyser dans l’heure qui suit le prélèvement et savoir répéter au moins 3 fois).
V 480Facteurs pronostiques
L’âge et l’état fonctionnel des patients au moment du diagnostic (mesuré par l’index de Karnofsky qui va de 0 à 100) constituent les facteurs pronostiques cliniques principaux dans les tumeurs cérébrales malignes.
Trois biomarqueurs tumoraux ont été associés à un pronostic favorable dans les gliomes diffus de l’adulte et contribuent à définir la prise en charge des patients :
- • la codélétion des chromosomes 1p et 19q;
- • la mutation du gène IDH (qui code l’enzyme isocitrate déshydrogénase intervenant dans le métabolisme énergétique de la cellule);
- • la méthylation du gène MGMT (méthyl-guanine méthyl-transférase), qui code pour une enzyme de réparation de l’ADN.
VI Principes généraux des traitements
A Chirurgie
La chirurgie des tumeurs cérébrales a fait des progrès considérables et la mortalité et la morbidité postopératoires sont devenues très faibles grâce au développement de techniques permettant le repérage des régions fonctionnelles à préserver (IRM fonctionnelle préopératoire, chirurgie en condition éveillée avec stimulation corticale et sous-corticale, neuronavigation, échographie pero-pératoire). L’exérèse chirurgicale, quand elle est possible, outre son intérêt diagnostique, permet de soulager immédiatement les symptômes d’HTIC, d’améliorer les déficits liés à une compression cérébrale et la tolérance des traitements postopératoires éventuels comme la radiothérapie et la chimiothérapie. Elle est curative lorsqu’elle conduit à une exérèse complète des méningiomes de bas grade et des gliomes de grade I et améliore considérablement la durée de vie des patients souffrant de tumeurs primitives de haut grade quand l’exérèse peut être totale ou subtotale. La chirurgie permet aussi de lever des obstacles à l’écoulement du LCS liés à l’obstruction tumorale par la réalisation, entre autres, d’une dérivation ventriculaire ou d’une ventriculostomie. En revanche, dans certaines tumeurs très infiltrantes et radio-chimio-sensibles comme les lymphomes primitifs du système nerveux central, l’exérèse chirurgicale n’apporte pas de bénéfice au traitement médical.
B Radiothérapie
La radiothérapie est le traitement médical de choix des tumeurs cérébrales de haut grade. Ses modalités varient selon la tumeur, son extension et le siège de rechute potentielle au sein du système nerveux. Elle doit prendre en compte le risque de neurotoxicité post-radique qui dépend de la dose totale, du fractionnement, du volume à irradier et des facteurs de risque liés au patient (âge, facteurs de risque cardiovasculaire), et la durée de survie attendue après le traitement.
 La dose par fraction optimale visant à réduire le risque de neurotoxicité post radique doit être < 1,8-2 Gy.
La dose par fraction optimale visant à réduire le risque de neurotoxicité post radique doit être < 1,8-2 Gy.
On distingue :
- • l’irradiation encéphalique totale : elle peut être discutée dans les métastases cérébrales multiples ou en consolidation dans les lymphomes cérébraux primitifs;
- • la radiothérapie externe focale conventionnelle : indiquée dans les gliomes diffus;
- • la radiothérapie conformationnelle : elle permet une collimation optimale de la lésion dans l’optique de limiter la dose des radiations délivrées aux structures cérébrales les plus fragiles (par exemple, le tronc cérébral ou les voies optiques);
- • la radiothérapie en condition stéréotaxique : elle repose sur l’administration en une séance d’une irradiation par de multiples faisceaux convergents (gamma knife – radiochirurgie 481ou cyberknife) et s’adresse à de petites lésions circonscrites (diamètre < 3 cm) comme les métastases cérébrales;
- • la radiothérapie craniospinale : pour les tumeurs à haut risque de dissémination au sein du névraxe et dans les méninges comme les médulloblastomes.
C Chimiothérapie
L’effet de la chimiothérapie dans le traitement des tumeurs cérébrales est limité par la barrière hémato-encéphalique (BHE), qui réduit son accès au parenchyme cérébral, et par la chimiorésistance intrinsèque de la majorité des tumeurs cérébrales primitives, en particulier les gliomes malins.
Les agents les plus efficaces sont des molécules de petites tailles et liposolubles. Outre son action cytotoxique propre, la chimiothérapie peut contribuer, pour certaines tumeurs comme le glioblastome, à accroître la sensibilité des cellules tumorales à la radiothérapie quand ces deux traitements sont administrés de façon concomitante. Les tumeurs primitives malignes les plus chimiosensibles sont les lymphomes, les germinomes, les oligodendrogliomes, les médulloblastomes.
D Corticothérapie
Les corticoïdes oraux (méthylprednisolone : Médrol®; prednisolone : Solupred®; prednisone : Cortancyl®) ou parentéraux (méthylprednisolone hémisuccinate : Solumédrol®) sont fréquemment utilisés en neuro-oncologie. Outre une action antitumorale propre (lymphome cérébral primitif), les corticoïdes agissent essentiellement sur l’œdème péritumoral. Ils permettent ainsi une réduction de l’HTIC et une amélioration fonctionnelle rapide (réduction des déficits et des crises comitiales).
Le problème essentiel des corticoïdes réside dans leurs effets indésirables. La prescription de corticoïdes doit donc toujours être revue de manière à ce qu’un patient donné reçoive seulement la dose minimale efficace adaptée à sa situation.
E Traitement antiépileptique
Un traitement antiépileptique est recommandé chez les patients ayant présenté une crise inaugurale ou continuant à souffrir de crises itératives. Sauf rares exceptions (métastases de mélanome), il n’est pas indiqué au long cours chez les patients dont l’histoire ne comporte aucune crise.
Le choix du traitement utilisé ne présente pas de spécificité; les médicaments les plus fréquemment employés en première intention sont des antiépileptiques non inducteurs enzyma-tiques et bien tolérés sur le plan cognitif :
- • par exemple : lévétiracétam (Keppra®), lacosamide (Vimpat®), lamotrigine (Lamictal®) en monothérapie.
En cas de chimiothérapie envisagée, les agents non inducteurs enzymatiques sont privilégiés. Ils nécessitent une surveillance des effets indésirables.
F Autres traitements symptomatiques
Divers traitements peuvent être nécessaires au cours de l’évolution d’une tumeur cérébrale de haut grade : antidépresseurs, anxiolytiques, anticoagulants, antalgiques, antiémétiques, protecteurs gastriques et autres.
G 482Soins palliatifs
Malgré un traitement optimal comprenant une exérèse chirurgicale, une radiothérapie et plusieurs lignes de chimiothérapie, la tumeur peut récidiver ou poursuivre son évolution. Une décision collégiale d’arrêt des traitements oncologiques pour poursuivre un traitement de confort peut alors être prise.
VII Particularités clinico-radiologiques et traitements spécifiques
Voir tableau 26.2 et encadré 26.1
Principales tumeurs cérébrales, traitement et pronostic.
| Tumeur | Traitement | Pronostic (médiane de survie) | |
|---|---|---|---|
| Gliomes (30 % des TCP) | Gliome de grade I « circonscrit » | Chirurgie | Guérison |
| Oligodendrogliome et astrocytome de grade II IDH muté (10 % des gliomes) | Chirurgie Si inopérable et signes évolutifs ou présence de facteurs de mauvais pronostic : RT + chimiothérapie adjuvante |
> 10 ans | |
| Oligodendrogliome astrocytome de grade III IDH muté (20 % des gliomes) |
Chirurgie + RT focale ± chimiothérapie dont les modalités dépendent du profil moléculaire | 5–10 ans | |
| Glioblastome (grade IV) IDH non muté (70 % des gliomes) | Chirurgie + RT focale + chimiothérapie par TMZ concomitant et adjuvant | 12–18 mois | |
| Méningiome (40 % des TCP) | Chirurgie | Guérison | |
| Lymphome primitif du SNC (5 % des TCP) | Chimiothérapie à base de MTX haute dose (chez les patients jeunes : ± RT encéphale in toto ou intensification avec autogreffe des cellules souches périphériques) | 3–4 ans Guérison : 30 % | |
| Médulloblastome (< 1 % TCP) | Chirurgie + RT craniospinale + chimiothérapie en cas de résection incomplète ou de diffusion leptoméningée | 5–10 ans | |
| Métastases cérébrales | Chirurgie Radiochirurgie (si < 3 et diamètre < 3 cm) RT de l’encéphale in toto Chimiothérapie si cancer chimio-sensible |
3–4 mois |
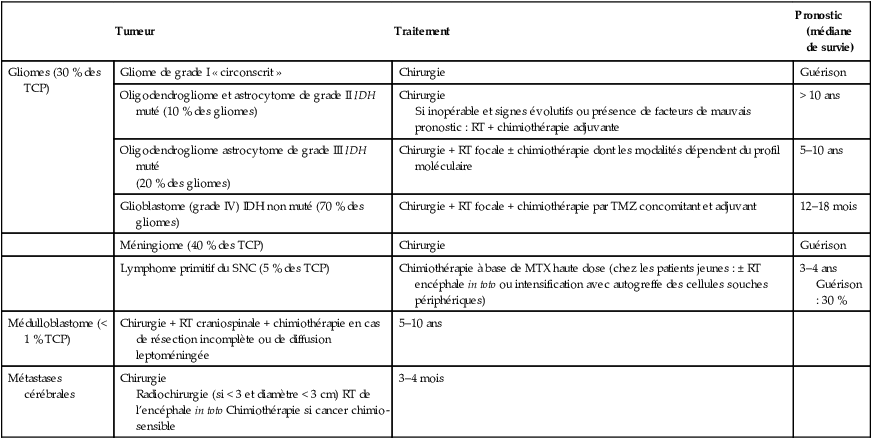
MTX : méthotrexate; RT : radiothérapie; SNC : système nerveux central; TCP : tumeur cérébrale primitive; TMZ : témozolomide.
A Gliomes
1 Gliomes circonscrits de grade I (exemple de l’astrocytome pilocytique)
a Particularités cliniques
Il s’agit d’une tumeur de l’enfant ou plus rarement de l’adulte jeune. Elle peut survenir isolément ou dans un contexte de maladie de von Recklinghausen ou neurofibromatose de type 1 (NF1).
b Aspects radiologiques
Il s’agit de tumeurs bien circonscrites, hypodenses en TDM, hyperintenses en IRM T2, prenant souvent le contraste et comportant souvent une composante kystique.
c Traitement
La guérison est la règle dès lors que les tumeurs sont résécables chirurgicalement. Dans les formes inopérables évolutives, une radiothérapie ou une chimiothérapie peuvent être proposées.
2 Gliome diffus de bas grade (astrocytome IDH muté et oligodendrogliome de grade II)
a Particularités cliniques
Les gliomes surviennent classiquement chez l’adulte jeune (30–40 ans) et se révèlent le plus souvent par une crise d’épilepsie. L’évolution naturelle est l’extension progressive de l’infiltrat tumoral de proche en proche et une transformation anaplasique en grade III ou IV, dont dépend alors le pronostic. Si la médiane de survie est de 5–10 ans, il existe une grande hétérogénéité évolutive en fonction du profil moléculaire de la tumeur, certains patients progressant rapidement vers la malignité et décédant en 2 ou 3 ans, d’autres ayant une tumeur relativement stable pendant de nombreuses années et compatible avec une vie longtemps normale. Du fait de l’évolution tumorale relativement lente qui induit d’importants phénomènes de plasticité cérébrale, les patients sont le plus souvent indemnes de déficit neurologique majeur, même en cas de tumeurs volumineuses. Toutefois, divers troubles cognitifs sont le plus souvent détectés après une évaluation neuropsychologique détaillée, recommandée chez ces patients dans le bilan préopératoire et le suivi.
b 483Aspects radiologiques
Les gliomes de grade II sont des tumeurs infiltrantes apparaissant en hyposignal T1, ne se rehaussant classiquement pas par le gadolinium, et en hypersignal T2 ou FLAIR. Au scanner, les tumeurs apparaissent hypodenses. Des calcifications intratumorales sont parfois visibles.
c Traitement
Une exérèse chirurgicale la plus complète possible est recommandée en cas de tumeur opérable.
Au vu de la localisation fréquente de ces tumeurs dans des zones ou à proximité de zones hautement fonctionnelles, cette exérèse est le plus souvent réalisée en condition éveillée. Cette technique permet la réalisation de cartographies fonctionnelles corticales et sous-corticales en temps réel et de détecter, et ainsi de préserver, les régions éloquentes. C’est le traitement recommandé en première intention pour les gliomes diffus de bas grade, car il permet une exérèse tumorale maximale selon des limites fonctionnelles, tout en préservant la qualité de vie des patients. Si la tumeur est inopérable, une biopsie pourra être discutée.
Une radiothérapie cérébrale focale sera discutée en cas d’inopérabilité et de signes d’évolu-tivité clinique ou radiologique de la tumeur ou de la présence de facteurs de mauvais pronostic (clinique ou moléculaire). Une chimiothérapie adjuvante (agents alkylants comme le témozolo-mide et les nitroso-urées) est proposée en complément de la radiothérapie.
Dans certaines tumeurs inopérables très étendues, pour lesquelles la radiothérapie nécessiterait des champs d’irradiation très larges exposant le patient à un risque accru de neurotoxicité, une chimiothérapie néoadjuvante est parfois proposée après discussion pluridisciplinaire. Une thérapie ciblée par vorasidénib ciblant IDH muté a aussi montré son efficacité, mais sa place reste à définir.
3 Gliomes diffus malins (oligodendrogliome et astrocytome IDH muté ID de grade III, glioblastome de grade IV)
a Particularités cliniques et radiologiques
Les gliomes dits de « haut grade » (III et IV) surviennent chez l’adulte plus âgé (âge moyen 50–60 ans). Ils peuvent survenir de novo ou provenir de la transformation maligne d’une tumeur de plus bas grade préexistante (gliomes malins secondaires ou « dégénérés »). À l’imagerie, les gliomes de haut grade sont d’aspect plus hétérogène, volontiers accompagnés d’un œdème et d’une prise de contraste.
b Traitement
Gliomes anaplasiques (astrocytome IDH muté et oligodendrogliome de grade III)
Le traitement des gliomes de grade III repose sur la chirurgie quand elle est possible et une radiothérapie focale sur le lit tumoral. Les tumeurs présentant une codélétion chromosomique 1p et 19q et/ou une mutation du gène de l’enzyme isocitrate déshydrogénase (IDH) sont chimiosensibles et justifient l’adjonction d’une chimiothérapie à base d’alkylants (nitroso-urées ou témozolomide) qui peut être délivrée juste avant (condition néoadjuvante) ou juste après (condition adjuvante) la radiothérapie. Pour les gliomes de grade III sans codélétion ni mutation du gène IDH, la séquence optimale de la chimiothérapie reste à déterminer.
Glioblastomes (grade IV, IDH non muté)
Les glioblastomes sont les tumeurs gliales les plus agressives. Le traitement standard repose sur la chirurgie, suivie d’une radiothérapie focale combinée de façon concomitante à une chimiothérapie par témozolomide, qui sera ensuite poursuivie en condition adjuvante par plusieurs 484cycles supplémentaires. La récidive locale est malheureusement la règle et la médiane de survie est de 16 mois. Récemment, un dispositif délivrant un champ électrique à basse fréquence (tumor treating fields ou TTF) à partir d’électrodes appliquées sur le cuir chevelu pendant la chimiothétapie adjuvante a montré améliorer la survie.
B Méningiome
1 Particularités cliniques
Les méningiomes sont des tumeurs le plus souvent bénignes qui se développent dans l’espace sous-dural aux dépens des cellules arachnoïdiennes. Le pic d’incidence se situe autour de la sixième décennie. Il existe une prédominance féminine avec un sex-ratio de 2:1. La plupart des méningiomes sont sporadiques, mais ils peuvent se développer dans le cadre d’une neurofibro-matose de type 2 (méningiomes multiples, association à des neurinomes). Leur découverte est souvent fortuite, car ils sont souvent asymptomatiques. S’ils sont cliniquement symptomatiques, le tableau dépend de la localisation de la tumeur. Ils peuvent se révéler par une crise d’épilepsie.
2 Aspects radiologiques
Le scanner sans et avec injection de produit de contraste a une excellente sensibilité pour détecter un méningiome. Les méningiomes sont fréquemment calcifiés. L’IRM est plus performante dans l’évaluation des rapports anatomiques de la tumeur, notamment vasculaires. L’aspect radiologique est typique : lésion homogène, très bien circonscrite, parfois polylobée, prenant le contraste de façon massive et homogène. Il existe une base d’implantation durale et, classiquement, un épaississement dural caractéristique en continuité avec le méningiome (« signe de la queue de comète ») est retrouvé.
3 Traitement
Le traitement repose sur la chirurgie.
Les indications opératoires dépendent des caractéristiques du méningiome (taille, localisation) et de son retentissement clinique. Il peut être tout à fait licite de proposer une surveillance simple devant une lésion asymptomatique. Une radiothérapie focalisée est discutée dans les méningiomes inopérables et évolutifs, dans les cas très rares de signes de malignité à l’analyse neuropathologique ou lors de récidive non opérable. Le méningiome étant une tumeur potentiellement hormonosensible, les traitements œstroprogestatifs sont par précaution à éviter. Le risque encouru doit néanmoins toujours être confronté au bénéfice du patient.
C Métastases cérébrales
1 Aspects radiologiques
Au scanner et à l’IRM, les métastases cérébrales apparaissent classiquement comme des lésions nodulaires prenant le contraste de façon variable (le plus souvent hétérogène mais parfois homogène ou annulaire). Elles sont le plus souvent accompagnées d’un œdème et d’un effet de masse important au regard de la taille des lésions. Elles sont volontiers hémorragiques dans le cas des mélanomes et, dans une moindre mesure, dans les cancers du rein. Elles siègent préférentiellement dans les zones de jonctions cortico-sous-corticales. L’IRM a une meilleure sensibilité que la TDM et permet de détecter des localisations passées inaperçues à la TDM. Les métastases cérébrales sont le plus souvent multiples mais peuvent être uniques dans 30 % des cas.
2 485Traitement
Le traitement des métastases cérébrales repose sur la chirurgie pour les lésions uniques, la radiothérapie stéréotaxique (radiochirurgie ou cyberknife) pour les lésions uniques ou multiples de taille < 3 cm, la radiothérapie de l’encéphale in toto dans les lésions multiples, et dans une moindre mesure la chimiothérapie (qui s’adresse aux métastases cérébrales de cancers chimio-sensibles, en particulier les cancers du poumon à petites cellules et les cancers du sein) et les thérapies ciblées quand il existe une cible actionnable. Ces derniers traitements peuvent se combiner. Le choix du traitement optimal se fait en réunion multidisciplinaire. Le pronostic est sombre, avec une médiane de survie de 5–8 mois; cependant la majorité des patients traités décèdent des complications de leur maladie systémique, souvent avancée au moment où les métastases cérébrales sont découvertes. Le traitement des métastases cérébrales doit être ainsi considéré comme un traitement palliatif visant à améliorer ou prévenir les symptômes neurologiques. Pour certains cancers cependant, tels que le cancer du sein, certains cancers du poumon et le cancer du rein, des survies longues après traitement des métastases cérébrales sont observées.
D Lymphomes cérébraux primitifs
1 Particularités cliniques
Le lymphome cérébral primitif est favorisé par l’existence d’une immunodépression (Sida, transplantation d’organe nécessitant un traitement immunosuppresseur), mais il survient néanmoins, dans la grande majorité des cas, chez l’adulte immunocompétent avec un pic de fréquence autour de 60 ans.
Dans 90 % des cas, le lymphome est de type B à grandes cellules.
2 Aspects radiologiques
Scanner et IRM montrent typiquement une ou des lésions volontiers périventriculaires se rehaussant de manière intense et homogène après injection de produit de contraste, prenant un aspect « cotonneux ». Chez le patient immunodéprimé, les lésions sont plus souvent multiples et prennent volontiers le contraste de façon annulaire, soulevant dans ce contexte le diagnostic différentiel d’une toxoplasmose cérébrale.
3 Traitement
Les lymphomes cérébraux primitifs sont souvent cortico-sensibles. Pour cette raison, quand le diagnostic est suspecté devant une imagerie souvent évocatrice, la prescription de corticoïdes doit être évitée si possible avant la biopsie.
Il s’agit d’une tumeur chimio- et radio-sensible et l’exérèse chirurgicale n’a pas de place dans le traitement. Celui-ci repose sur une chimiothérapie à base de méthotrexate intraveineux à hautes doses qui permet à l’agent de passer la BHE. Le traitement peut être consolidé chez les sujets jeunes par une intensification de chimiothérapie avec greffe de cellules souches. De préférence à une radiothérapie encéphalique qui expose les patients à des séquelles neu-rocognitives (leucoencéphalopathie post-radique). Une rémission est obtenue dans la majorité des cas, mais les rechutes sont fréquentes et on estime que 20 à 30 % de guérisons peuvent être obtenues.
E 486Médulloblastome
1 Aspect clinique et radiologique
Il s’agit d’une tumeur de l’enfant et près de 70 %% des cas surviennent avant 20 ans. Typiquement, ces tumeurs intéressent le cervelet et se manifestent par une ataxie cérébelleuse et une HTIC par hydrocéphalie secondaire.
L’IRM montre typiquement une masse homogène en hypersignal T2 et isosignal T1, se rehaussant de façon relativement homogène. Le médulloblastome peut disséminer très précocement dans les méninges et beaucoup plus rarement hors du système nerveux central (métastases osseuses principalement). Le bilan d’extension doit ainsi comporter en postopératoire une IRM craniospinale et une étude du LCS.
2 Traitement
Le traitement standard repose sur la chirurgie et l’association chirurgie–radiothérapie. L’irradiation doit être craniospinale en raison du risque de dissémination méningée et de facteurs biologiques de mauvais pronostic. La place de la chimiothérapie est discutée en cas d’exérèse incomplète et/ou d’une dissémination méningée ou systémique. Dans les formes à faible risque de récidive d’après le profil moléculaire, des protocoles de chimiothérapie sont établis et la radiothérapie est parfois différée.
F Tumeurs hypophysaires
O Voir l’item 244 du réferentiel d’endocrinologie.
1 Adénomes hypophysaires
Les adénomes hypophysaires sont classés en adénomes non sécrétants (25 % des cas) et sécrétants. Ces derniers sont également divisés selon l’hormone synthétisée : prolactine, hormone de croissance (adénome somatotrope), LH et FSH (gonadotrope), ACTH (corticotrope), TSH (thyréotrope).
a Particularités cliniques
Les signes cliniques suivants, parfois associés, doivent faire rechercher une lésion hypophysaire :
- • le syndrome endocrinien d’hypersécrétion hormonale, par exemple aménorrhée-galactorrhée, ou d’hyposécrétion hormonale; dans quelques rares cas, cette dernière peut être aiguë et menacer le pronostic vital par la décompensation d’une insuffisance surrénalienne;
- • le syndrome tumoral avec signes neurologiques (HTIC, baisse de l’acuité visuelle et déficit campimétrique par compression des voies optiques, atteinte des paires crâniennes par envahissement du sinus caverneux, etc.).
b Diagnostic
Bilan hormonal
Le bilan hormonal cherche, en fonction du tableau clinique et de la taille de l’adénome, une hypersécrétion hormonale et/ou une insuffisance antéhypophysaire.
Imagerie
Le diagnostic repose sur une IRM comportant des coupes millimétriques centrées sur la selle turcique dans les trois plans de l’espace avec injection de gadolinium. L’examen permet de 487visualiser la tumeur (généralement en isosignal T1, en hypersignal T2, se rehaussant après injection) et d’évaluer ses rapports avec les structures de voisinage, en particulier le chiasma optique, le sinus caverneux et les artères carotides. Ces informations sont capitales en vue d’un geste chirurgical. Un examen ophtalmologique avec champ visuel sera systématiquement demandé.
c Traitement
Le traitement dépend du type de l’adénome. Globalement, il est chirurgical en première intention, sauf pour l’adénome à prolactine pour lequel un traitement médical par agoniste dopaminergique (bromocriptine, cabergoline) est d’abord proposé.
488489490491492493Les déficits endocriniens associés sont traités par hormonothérapie substitutive adaptée à chaque cas.