Situations de départ
 62 Troubles de déglutition ou fausse route.
62 Troubles de déglutition ou fausse route.
74 Faiblesse musculaire.
121 Déficit neurologique sensitif et/ou moteur.
143 Diplopie.
Objectifs pédagogiques
Diagnostiquer une myasthénie.
Connaître les principes de la prise en charge.
Identifier les situations d’urgence.
Hiérarchisation des connaissances
| Rang | Rubrique | Intitulé | Descriptif |
|---|---|---|---|
 |
Éléments physiopathologiques | Connaître les principaux éléments physiopathologiques de la myasthénie | |
 |
Diagnostic positif | Savoir diagnostiquer une myasthénie | Connaître les symptômes révélateurs les plus fréquents et les plus évocateurs (oculaires, bulbaires, muscles respiratoires) avec fatigabilité ou variabilité dans le temps |
|
Diagnostic positif | Connaître et savoir rechercher les pathologies fréquentes associées à la myasthénie | Pathologie thymique, pathologies auto-immunes |
|
Diagnostic positif | Connaître l’existence de formes oculaires pures et de formes généralisées de myasthénie | |
|
Examens complémentaires | Connaître les éléments paracliniques du diagnostic | Électroneuromyogramme, anticorps antirécepteurs à l’acétylcholine |
|
Identifier une urgence | Reconnaître les situations d’urgence de la myasthénie | Crises myasthéniques, infections, médicaments |
|
Identifier une urgence | Connaître les risques et les complications graves de la myasthénie | |
|
Prise en charge | Planifier la prise en charge des situations d’urgence | |
|
Prise en charge | Connaître les principes du traitement | Traitement symptomatique (anticholinergique), traitement de fond, médicaments prohibés |
157 La myasthénie (ou myasthenia gravis) :
- • la myasthénie est une maladie auto-immune liée à un blocage des récepteurs de la plaque motrice par des anticorps antirécepteurs de l’acétylcholine ou d’autres types d’anticorps induisant un dysfonctionnement de la transmission neuromusculaire : il s’agit d’un bloc post-synaptique;
- • la responsabilité du thymus est importante : les récepteurs de l’acétylcholine des cellules myoïdes du thymus entraînent la stimulation d’anticorps contre les récepteurs de la jonction neuromusculaire; le thymus est une source de lymphocytes T helper stimulant la production de ces anticorps par les lymphocytes B;
- • elle peut survenir à tout âge, mais atteint le plus souvent les adultes;
- • elle est plus fréquente chez la femme avant 50 ans, alors qu’au-dessus de 60 ans, la myasthénie prédomine chez les hommes.
I Diagnostic clinique
Le diagnostic de myasthénie doit être évoqué devant des signes et symptômes, conséquences d’une fatigabilité excessive de la musculature striée à l’effort et fluctuant dans le temps (fatigabilité ou phénomène myasthénique) qui le plus souvent :
- • apparaît ou augmente à l’effort;
- • augmente en fin de journée;
- • se corrige au repos;
- • peut se manifester dans les muscles directement mis en action au cours de l’effort;
- • peut associer des signes évocateurs : ophtalmoplégie (ptosis et diplopie), faiblesse musculaire des membres, troubles de la déglutition.
A Muscles oculaires et palpébraux
- • Dans 50 % des cas, les premières manifestations sont purement oculaires, mais d’autres territoires sont affectés dans plus de 80 % des cas après 1 an d’évolution (myasthénie généralisée).
-
• Ptosis unilatéral au début, qui peut se bilatéraliser par la suite; il reste habituellement asymétrique (figure 9.1).
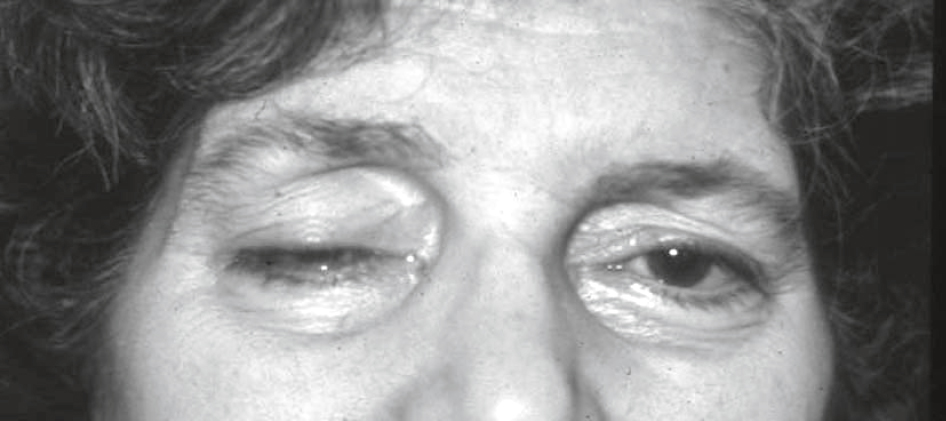
Fig. 9.1 Ptosis unilatéral droit.L'œil droit du patient montre une dissipation des yeux évidente. Ceci est caractérisé par une goutte proéminente de la paupière supérieure, couvrant partiellement l'iris et limitant l'ouverture du palais. En revanche, l'œil gauche est normalement ouvert, avec une symétrie faciale étant conservée dans le reste du visage. Un aspect général suggère la participation d'un côté isolé du corps sans d'autres signes visibles de paralysie ou d'anomalies de mouvement oculaire. Le front droit semble être légèrement abaissé, mettant en évidence l'asymétrie des paupières. La peau légèrement pliée autour des yeux indique l'âge adulte progressif. Le contraste entre la qualité de l'éclairage et du noir et du blanc met en évidence la différence d'ouverture entre les yeux.
- • Ptosis à bascule ou alternant quasiment pathognomonique.
- • Diplopie, le plus souvent intermittente, disparaissant en vision monoculaire.
- • Ptosis et diplopie sont augmentés par la fatigue, la lumière, la fixation d’un objet.
- • La diplopie est souvent difficile à analyser lorsque plusieurs nerfs oculomoteurs sont touchés (absence de systématisation contrairement aux atteintes des nerfs oculomoteurs).
- • La musculature pupillaire est indemne.
B 158Muscles d’innervation bulbaire
- • Troubles de la déglutition, de la phonation et de la mastication.
- • La voix s’éteint progressivement, devient nasonnée puis inintelligible.
- • Troubles de la mastication qui apparaissent au cours des repas, le sujet se trouvant parfois dans l’obligation de soutenir sa mâchoire inférieure avec sa main.
- • Troubles de la déglutition avec fausses routes et parfois reflux des liquides par le nez lorsque coexiste une atteinte du voile du palais.
- • Troubles de la motricité linguale (difficultés de propulsion linguale).
- • Une parésie faciale donnant un faciès atone est souvent associée aux troubles bulbaires (sourire vertical).
C Autres muscles
1 Muscles des membres
L’atteinte prédomine sur les muscles proximaux, mais peut aussi toucher la musculature distale (plus rarement).
2 Muscles axiaux
- • Atteinte des muscles abdominaux entraînant des difficultés pour se relever du décubitus.
- • Fatigabilité des muscles cervicaux à l’origine d’une chute de la tête en avant « tête tombante ») et de douleurs cervicales.
D Muscles respiratoires
- • L’atteinte respiratoire peut se traduire par une dyspnée à l’effort puis au repos, une ortho-pnée, une toux inefficace.
- • Elle peut survenir très soudainement et conduire à une insuffisance respiratoire aiguë, engageant le pronostic vital (+++).
E 159Examen clinique
- • Il permet de constater les signes évocateurs de myasthénie lorsqu’ils sont présents : ptosis, paralysies oculomotrices non systématisées, hypotonie faciale, chute de la nuque et de la mâchoire.
- • Le ptosis peut être majoré après fixation du regard vers le haut et amélioré après un test au glaçon (dans une poche en plastique appliquée 2 minutes sur l’œil fermé).
- • Il peut mettre en évidence une fatigabilité anormale des membres lors des épreuves de Barré et de Mingazzini qui sont normalement tenues 2,30 minutes et 1,15 minute respectivement.
- • Absence de trouble sensitif, d’anomalie des réflexes, et absence de signe d’atteinte neurologique centrale.
F Évolution
- • Chronique et imprévisible, avec périodes de poussée et de rémission, et une tendance à l’aggravation dans les premières années : pour 85 % des patients, le stade de gravité maximum (risque de généralisation et de poussées graves) est atteint dans un délai inférieur à 3 ans.
- • Complications vitales possibles lors des crises myasthéniques, se manifestant par des troubles de la déglutition et respiratoires, avec dyspnée et encombrement. Les crises sévères nécessitent une hospitalisation en réanimation.
- • Possible aggravation de la myasthénie au cours de la grossesse et en post-partum.
G Reconnaître les situations d’urgence de la myasthénie
- • L’atteinte des muscles respiratoires et les troubles sévères de déglutition sont susceptibles d’engager le pronostic vital et caractérisent les formes graves (20 à 30 % des patients).
- • Les symptômes devant alerter sont les suivants : encombrement, essoufflement au moindre effort, orthopnée, toux non efficace, fausses routes et/ou une faiblesse musculaire devenant très marquée.
- • L’aggravation rapide sur quelques jours, voire quelques heures, des symptômes fait craindre une crise myasthénique et nécessite une hospitalisation en urgence, éventuellement en service de réanimation.
-
• Si les symptômes sont rapides et sévères, l’appel du SAMU est indiqué :
-
– une aggravation motrice et respiratoire rapide peut également survenir au cours de la crise cholinergique, parfois difficile à distinguer de la crise myasthénique :
- – elle est due à un surdosage en anticholinestérasiques,
- – elle se caractérise par la présence de signes de surdosage cholinergique : fascicu-lations diffuses, signes digestifs (nausées, vomissements, diarrhée), hypersalivation, hypersécrétion bronchique, myosis, bradycardie,
- – elle est beaucoup plus rare que la crise myasthénique, mais les deux complications peuvent s’intriquer,
- – le passage en réanimation s’impose.
-
II 160Éléments paracliniques du diagnostic
A Recherche des autoanticorps
-
• Anticorps antirécepteurs de l’acétylcholine (anti-RACh) :
- – présents chez 80 % des malades avec myasthénie généralisée et chez 50 % de ceux avec myasthénie oculaire (voir plus loin);
- – absence de corrélation entre le taux d’anticorps et la gravité de la maladie;
- – en revanche, chez un même sujet, le taux peut fluctuer en fonction de l’évolutivité de la maladie;
- – dans les thymomes malins, le taux est souvent très élevé.
- • Anticorps anti-MuSK (dirigés contre une protéine post-synaptique associée au récepteur à l’acétylcholine : Muscle Spécifie Kinase) : 10 % environ des myasthénies généralisées.
- •
 Anticorps anti-LRP4 et anticorps anti-RACh à faible affinité (5 à 10 %% des formes généralisées).
Anticorps anti-LRP4 et anticorps anti-RACh à faible affinité (5 à 10 %% des formes généralisées).
B Recherche de décrément en ENMG (stimulodétection répétitive)
L’examen électrophysiologique permet de mettre en évidence un bloc neuromusculaire. Le nerf moteur est stimulé dix fois de suite à la fréquence de 3 Hz et l’amplitude de la réponse musculaire (potentiel moteur) est enregistrée.
Dans le syndrome myasthénique, la perturbation de la transmission neuromusculaire entraîne une diminution de l’amplitude du potentiel moteur (décrément) qui est supérieure à 10 % (figure 8.2).

Décrément à l’ENMG. Diminution de l’amplitude du potentiel évoqué musculaire lors des stimulations répétées du nerf à basse fréquence.
Plusieurs tracés électromyographiques successifs sont représentés ici, illustrant un décrément progressif de l’amplitude du potentiel moteur. Chaque courbe présente une réponse musculaire évoquée par une série de stimulations répétées à fréquence constante, dont l’amplitude diminue progressivement, phénomène typique observé dans certains troubles de la transmission neuromusculaire comme la myasthénie. La première onde à gauche est marquée par une amplitude plus élevée, suivie de réponses successives qui décroissent de façon visible avant de se stabiliser. Ce schéma traduit une diminution de la réponse post-synaptique, suggérant une défaillance progressive du couplage neuromusculaire au cours des stimulations répétées. L’image met ainsi en évidence un signe classique de défaillance synaptique dans un contexte de pathologie neuromusculaire, observable à l’ENMG.
La recherche de décrément doit être effectuée si possible en l’absence de traitement anti-cholinestérasique (arrêt depuis plus de 24 h), au niveau de plusieurs couples nerf–muscle comme les nerfs facial, spinal, ulnaire, radial, en privilégiant les territoires symptomatiques.
La sensibilité diagnostique ne dépasse pas 75 %.
Lorsque le bloc manque, on pourra pratiquer un examen sur fibre unique, plus complexe à réaliser, effectué uniquement dans quelques centres, qui peut mettre en évidence un allongement du jitter (variation du temps de réponse d’une seule fibre musculaire après stimulation du nerf, ou variation entre la réponse de deux fibres musculaires d’une même unité motrice lors d’un effort volontaire).
C 161Test pharmacologique aux anticholinestérasiques
- • À pratiquer en hospitalisation seulement, par crainte d’un syndrome vagotonique ou d’une crise cholinergique (voir plus loin).
- • Administration d’édrophonium IV (Enlon®) par voie intraveineuse lente (1 ampoule à 10 mg) ou de néostigmine (Prostigmine®) par voie sous-cutanée ou intramusculaire (0,5 mg), associée à une injection de 0,25 mg d’atropine pour éviter les effets secondaires intestinaux et une bradycardie.
- • L’amélioration ou la disparition des signes neurologiques objectivables (ptosis, déficit moteur des muscles d’innervation bulbaire, faiblesse des membres), si elle est franche et rapide (délai d’action < 5 minutes pour l’édrophonium, et de 15 minutes à 1 heure pour la néostigmine), a un grand intérêt diagnostique.
D Imagerie
Le scanner thoracique sans injection doit être systématique devant toute suspicion de myasthénie, afin d’explorer la loge thymique à la recherche d’une hyperplasie thymique ou d’un thymome (bénin ou malin) (figure 9.3).
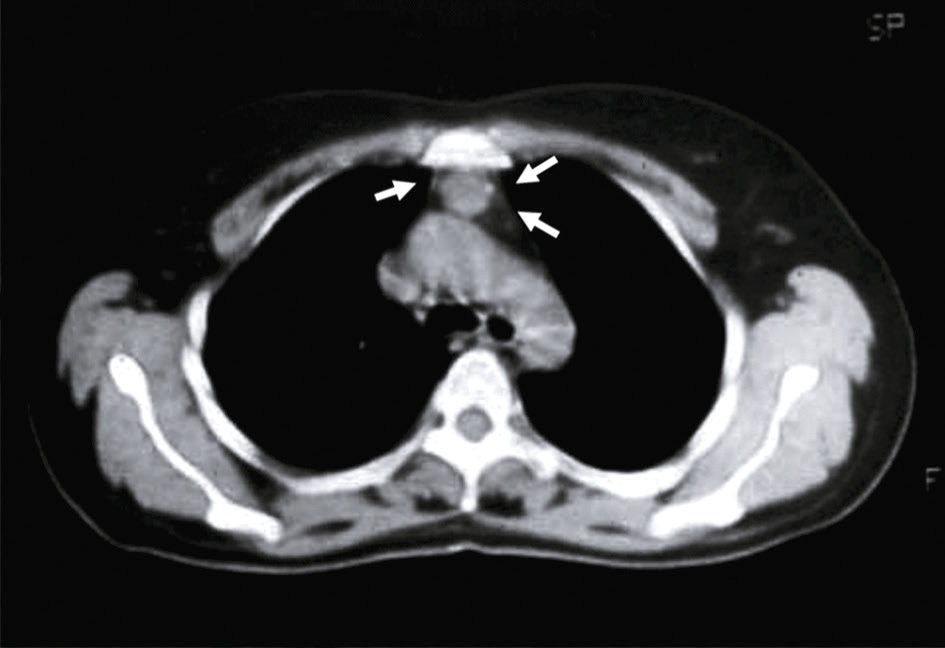
Scanner thoracique. Thymus hyperplasique résiduel.Sur cette coupe axiale de scanner thoracique injecté, on observe une masse tissulaire triangulaire en densité homogène située en avant des gros vaisseaux du médiastin antérieur. Les flèches blanches désignent cette structure bien délimitée, en position rétro-sternale haute. Son rehaussement modéré et sa symétrie par rapport aux lignes médianes suggèrent un thymus hyperplasique résiduel, souvent retrouvé chez l’adulte jeune ou après un stress systémique. Aucun signe d’infiltration des structures vasculaires adjacentes ni de masse compressive n’est observé. L’aspect global oriente vers une hyperplasie thymique bénigne plutôt qu’une pathologie tumorale invasive.
En cas de doute sur un thymome, une injection d’iode est indiquée, mais en dehors d’une poussée, car elle peut aggraver une myasthénie instable.
La négativité du bilan ne rejette pas formellement le diagnostic, et il faudra repérer le dosage des anticorps anti-Rach et anti-MuSK quelques mois après le premier dosage en raison d’une positivation secondaire possible.
III Pathologies associées
A Thymus et myasthénie
- • Chez 60 % des patients, en majorité des femmes de moins de 45 ans avec des anticorps anti-RACh, le thymus est le siège d’une hyperplasie (prolifération de follicules germinatifs composés de lymphocytes B avec une couronne de lymphocytes T).
- • Quinze à 20 % des patients présentent un thymome (tumeur constituée de cellules épi-théliales et lymphocytaires), habituellement après 40 ans. Les thymomes peuvent être 162bénins (absence de franchissement de la capsule thymique) ou malins, et doivent être opérés.
- • La présence d’anticorps anti-RACh est quasi constante en cas de thymome.
- • Une fois le thymome traité, la mortalité reste faible même si le thymome est invasif.
B Myasthénie et maladies auto-immunes
- • L’association de la myasthénie à d’autres maladies auto-immunes est possible.
- • Affection thyroïdienne le plus souvent (maladie de Basedow, thyroïdite) : environ 10 % des patients.
- • Autres associations, dans environ 5 % des cas : polyarthrite rhumatoïde, syndrome de Gougerot-Sjögren, maladie de Biermer, lupus érythémateux disséminé, etc.
- • Le bilan biologique à la recherche d’autres affections auto-immunes comportera les dosages suivants : thyroxine (T4), thyroid stimulating hormone (TSH), anticorps antithyroïdiens, dosage de vitamine B12, facteurs rhumatoïdes, facteur antinucléaire (FAN), anticorps anti-DNA et anti-ENA.
IV Formes cliniques
A Formes oculaires de myasthénie
- • Chez 10 à 15 % des patients, l’atteinte reste localisée aux muscles oculaires après 2 ans, et l’on parle alors de myasthénie oculaire, même si dans de rares cas une généralisation peut survenir plus tardivement.
- • Il faut distinguer la myasthénie oculaire des symptômes oculaires qui sont très fréquents au début de la maladie (50 % des cas) et le plus souvent suivis d’une généralisation. On ne peut donc pas poser le diagnostic de myasthénie oculaire avant un recul de 2 ans après les premiers signes.
-
• Les caractéristiques de la myasthénie oculaire sont les suivantes :
- – prédominance masculine;
- – survenue le plus souvent après 40 ans;
- – rareté du thymome;
- – fréquente absence de bloc neuromusculaire à l’ENMG (car recherché sur des couples nerfs–muscles extra-oculaires);
- – anticorps anti-RACh absents dans la moitié des cas;
- – mauvaise réponse au traitement anticholinestérasique.
- • Ces formes de myasthénie posent essentiellement un problème fonctionnel.
B Formes avec anti-MuSK
- • Prépondérance chez les femmes et, le plus souvent, formes généralisées.
- • Atteinte bulbaire plus fréquente que dans les autres formes, avec atrophie de la langue.
- • Fréquence des crises myasthéniques avec atteinte respiratoire.
- • Absence de thymome.
- • Fréquente négativité de l’exploration ENMG.
- • Mauvaise réponse aux anticholinestérasiques.
- • 163Meilleure efficacité des échanges plasmatiques par rapport aux Ig IV.
- • Très grande efficacité des anti-CD20 (rituximab).
C Myasthénie et grossesse
- • Risqué élevé d’aggravation de la myasthénie au cours du 1er trimestre, et dans les jours ou semaines qui suivent l’accouchement (30 à 40 % des cas).
- • Dix à 20 % des nouveau-nés de mère myasthénique présentent une myasthénie néonatale.
- • Il n’existe aucune corrélation entre la sévérité de la myasthénie chez la mère et la survenue ou la gravité de la myasthénie néonatale.
D Myasthénie néonatale
- • Transitoire, elle survient chez 10 à 20 % des nouveau-nés de mère myasthénique.
- • Elle est due à un transfert transplacentaire passif des anticorps maternels anti-RACh.
- • Les symptômes se manifestent très précocement, de quelques heures à 3 jours après l’accouchement.
- • Habituellement légère, elle se manifeste par une hypotonie associée à des troubles de la succion, mais elle peut être sévère avec des troubles respiratoires et de la déglutition.
- • Après prise en charge, l’évolution est toujours favorable avec régression complète de la myasthénie dans les 3 premiers mois de vie (par disparition des anticorps maternels).
V Traitements
A Traitement symptomatique
- • Les anticholinestérasiques sont prescrits en première intention, permettant d’améliorer transitoirement les symptômes musculaires.
- • Ils prolongent l’action de l’acétylcholine au niveau de la membrane post-synaptique par blocage réversible de l’acétylcholinestérase.
-
• Deux produits sont disponibles par voie orale :
- – la pyridostigmine (Mestinon®), comprimés à 60 mg, dont l’action est d’environ 4 heures;
- – l’ambénonium (Mytelase®), comprimés à 10 mg, dont l’effet est plus long (4 à 6 heures).
- • La posologie quotidienne sera augmentée progressivement jusqu’à la dose optimale, à adapter à chaque patient en fonction de son activité et des moments de plus grande fati-gabilité (6 à 8 comprimés maximum par jour répartis en 3 à 4 prises).
- • Efficacité moindre voire intolérance dans les formes avec anticorps anti-MuSK.
- • Il existe une forme retard de la pyridostigmine, pouvant être administrée au coucher, lorsque les symptômes sont présents dès le réveil (troubles de la déglutition notamment).
- • Effets indésirables en rapport avec les effets muscariniques (diarrhées, douleurs abdominales, hypersalivation, hypersécrétion bronchique, sueurs, bradycardie) ou nicotiniques (fasciculations, crampes).
- • Risque de la survenue d’une crise cholinergique en cas de surdosage, en particulier chez les patients dont la myasthénie se décompense : hypersécrétion bronchique, accentuation de la faiblesse musculaire, fasciculations et crampes musculaires.
B 164Thymectomie
- • Elle est toujours indiquée en cas de thymome, quelle que soit la sévérité de la myasthénie.
- • Cette intervention ne doit toutefois jamais être effectuée en urgence, et jamais chez un patient en poussée de myasthénie.
- • Elle doit également être discutée chez les patients de moins de 60 ans ayant des anticorps anti-RACh, particulièrement dans les myasthénies récentes (< 3 ans).
- • Son bénéfice est démontré, mais l’effet est retardé (> 6 mois après l’intervention).
- • Elle n’est pas indiquée dans les formes avec anticorps anti-MuSK.
C Corticothérapie
- • La prednisone est le plus souvent utilisée et est prescrite à la dose initiale de 1 mg/kg par jour dans la myasthénie généralisée, pendant 4 à 6 semaines. Les posologies seront progressivement réduites après amélioration significative et stabilisation (jusqu’à 0,5 mg/kg par jour à la fin du 4e mois, puis 0,25 mg/kg par jour après 9 mois).
- • La mise en route peut se faire en milieu hospitalier en raison d’un risque d’aggravation transitoire dans les 2 premières semaines de traitement (ou avec une introduction à doses progressives), mais la réponse est le plus souvent rapide au cours du 1er mois de traitement.
- • Elle peut être prescrite dans les myasthénies oculaires rebelles aux anticholinestérasiques à la posologie de 0,5 mg/kg par jour.
- • La prescription prolongée des corticoïdes expose le patient aux risques d’effets secondaires classiques de la corticothérapie.
D Autres immunosuppresseurs
- • L’azathioprine est l’immunosuppresseur le plus souvent prescrit, à la posologie initiale de 2 à 3 mg/kg/jour (100 ou 150 mg par jour selon le poids). Son effet est toujours retardé, après 6 semaines à 3 mois.
- • Un dosage de l’activité thymidine phosphorylase lymphocytaire (TPMT) est souhaitable avant l’instauration du traitement en raison d’une toxicité hématologique accrue chez les patients ayant un déficit en TPMT.
- • Les effets secondaires hématologiques (leucopénie, thrombocytopénie) et digestifs (hépatite, pancréatite) nécessitent une surveillance biologique étroite hebdomadaire le premier mois, puis mensuelle.
- • L’azathioprine est le plus souvent prescrit en association à la corticothérapie en début de traitement (3 premiers mois), en raison d’une potentialisation de leurs effets et pour permettre une épargne cortisonique plus rapide.
- • Le traitement ne doit être arrêté qu’après plusieurs années de stabilisation de la myasthénie, afin de limiter les risques de rechute.
- • Le mycophénolate mofétil (Cellcept®) est une alternative à l’azathioprine, avec une efficacité voisine, mais est formellement contre-indiqué en cas de grossesse.
- • D’autres immunosuppresseurs sont efficaces, mais leurs effets secondaires plus importants en limitent l’indication aux formes sévères, cortico-résistantes, ou après échec des immunosuppresseurs de première ligne. Le plus utilisé est le rituximab (MabThera®) – anticorps monoclonal anti-CD20 – dont l’efficacité est souvent très importante dans les myasthénies anti-MuSK.
- • 165De nouveaux traitements par anticorps monoclonaux – inhibiteurs de la fraction C5 du complément et inhibiteurs du FcRn – ont montré leur efficacité dans les formes réfractaires de myasthénie, et permettent de réduire la gravité des poussées sévères de myasthénie. Leur place dans la stratégie thérapeutique n’est pas encore clairement établie.
E Médicaments contre-indiqués
Tout patient doit porter sur lui une carte de myasthénie et la liste des principaux médicaments interdits (encadré 9.1).
VI Planifier le suivi des patients
-
•
La stratégie thérapeutique dépend des paramètres suivants :- – l’âge du patient;
- – l’existence ou non d’un thymome;
- – la gravité de la myasthénie (troubles de la déglutition ou respiratoires);
- – la tolérance des traitements et les risques thérapeutiques;
- – l’impact socioprofessionnel de la maladie.
-
• La gravité sera évaluée en tenant compte à chaque visite du score myasthénique de Garches (encadré 9.2
 ), du score d’activité quotidienne des 8 derniers jours (score Myasthenia Gravis Activities of Daily Living ou MG-ADL : Cf. Flashcode ci-contre) et de la gravité respiratoire (mesure de la capacité vitale en cas de troubles respiratoires, dyspnée, toux inefficace, etc.).
), du score d’activité quotidienne des 8 derniers jours (score Myasthenia Gravis Activities of Daily Living ou MG-ADL : Cf. Flashcode ci-contre) et de la gravité respiratoire (mesure de la capacité vitale en cas de troubles respiratoires, dyspnée, toux inefficace, etc.).
- • Traitement anticholinestérasique prescrit chez tous les patients, avec respect des contre-indications médicamenteuses (attention au risque d’aggravation des symptômes de myasthénie associés aux symptômes cholinergiques en cas de surdosage).
- • Thymectomie systématique en cas de thymome et indiquée dans les formes généralisées avec anticorps anti-RACh avant 60 ans (en dehors d’une poussée).
- • Si la myasthénie est sévère, la corticothérapie et/ou l’azathioprine seront prescrites en première intention au cours d’une hospitalisation.
- • 166En cas de poussée sévère : mesures de réanimation (sonde gastrique, assistance ventila-toire) en association à des plasmaphérèses ou des immunoglobulines IV, parallèlement à la prescription d’un traitement immunosuppresseur.
- • Éducation du patient et de son entourage aux signes d’alerte évocateurs d’une poussée, et nécessitant de se rendre aux urgences ou d’appeler le SAMU.
VII Identifier les situations d’urgence et planifier leur prise en charge
- • Des poussées aiguës de myasthénie (crise myasthénique) peuvent survenir spontanément ou être déclenchées par une prise de médicament contre-indiqué, une infection, une intervention chirurgicale, le post-partum.
- • Les signes majeurs de gravité sont l’encombrement, l’essoufflement au moindre effort avec orthopnée, la toux inefficace, les fausses routes et/ou une faiblesse musculaire devenant très marquée.
- • Une aggravation rapide de ces symptômes sur quelques jours, voire quelques heures, impose une hospitalisation d’urgence en service de réanimation. Si les symptômes sont rapides et sévères au domicile du patient, l’appel du SAMU est indiqué.
- • La pose de sonde gastrique et une intubation en urgence sont parfois nécessaires.
-
• Deux types de traitements sont utiles dans les poussées sévères, avec une efficacité semblable :
- – les immunoglobulines polyvalentes intraveineuses (Ig IV), en perfusion de 1 g/kg ou en cure de 2 g/kg sur 5 jours;
- – les échanges plasmatiques (EP) : de deux à quatre sur 1 à 3 semaines.
- • Le choix entre les Ig IV et les EP dépend de leurs contre-indications respectives, des possibilités de l’hôpital et de la réponse du patient à ces traitements lors d’une éventuelle poussée antérieure.
- • Ces traitements seront toujours associés à un traitement de fond, en raison de leur effet transitoire.
-
• Une aggravation motrice et respiratoire rapide peut également survenir au cours de la crise cholinergique, parfois difficile à distinguer de la crise myasthénique :
- – elle est due à un surdosage en anticholinestérasiques;
- – elle se caractérise par la présence de signes de surdosage cholinergique : fasciculations diffuses, signes digestifs (nausées, vomissements, diarrhée), myosis, bradycardie;
- – les deux complications peuvent s’intriquer;
- – le passage en réanimation s’impose.
167