104Situations de départ
Symptômes et signes cliniques
 2 Diarrhée.
2 Diarrhée.
36 Douleur de la région lombaire.
62 Troubles de déglutition ou fausse route.
64 Vertige et sensation vertigineuse.
65 Déformation rachidienne.
66 Apparition d’une difficulté à la marche.
71 Douleur d’un membre (supérieur ou inférieur).
72 Douleur du rachis (cervical, dorsal ou lombaire).
73 Douleur, brûlure, crampes et paresthésies.
74 Faiblesse musculaire.
77 Myalgies.
121 Déficit neurologique sensitif et/ou moteur.
127 Paralysie faciale.
130 Troubles de l’équilibre.
146 Dysphonie.
159 Bradycardie.
160 Détresse respiratoire aiguë.
162 Dyspnée.
166 Tachycardie.
105Données paracliniques
183 Analyse du liquide cérébrospinal (LCS).
Prise en charge aiguë et chronique
246 Prescription d’un soin ambulatoire.
247 Prescription d’une rééducation.
249 Prescrire des anti-inflammatoires non stéroïdiens (AINS).
250 Prescrire des antalgiques.
259 Évaluation et prise en charge de la douleur aiguë.
260 Évaluation et prise en charge de la douleur chronique.
271 Prescription et surveillance d’une voie d’abord vasculaire.
276 Prise en charge d’un patient en décubitus prolongé.
277 Consultation de suivi d’un patient présentant une lombalgie aiguë ou chronique.
Prévention
319 Prévention du surpoids et de l’obésité.
324 Modification thérapeutique du mode de vie (sommeil, activité physique, alimentation…).
Situations diverses
327 Annonce d’un diagnostic de maladie grave au patient et/ou à sa famille.
339 Prescrire un arrêt de travail.
342 Rédaction d’une ordonnance/d’un courrier médical.
352 Expliquer un traitement au patient (adulte/enfant/adolescent).
Objectifs pédagogiques
Item 95 – Radiculalgie et syndrome canalaire
Savoir diagnostiquer une radiculalgie et un syndrome canalaire.
Identifier les situations d’urgence et planifier leur prise en charge.
Item 96 – Neuropathies périphériques
Connaître les différentes formes de neuropathies périphériques et connaître l’orientation étiologique.
Connaître l’urgence du diagnostic et de la prise en charge des mononeuropathies multiples.
Item 97 – Polyradiculonévrite aiguë inflammatoire (syndrome de Guillain-Barré)
Diagnostiquer un syndrome de Guillain-Barré.
Identifier les situations d’urgence et connaître les grands principes de prise en charge.
Hiérarchisation des connaissances
Item 95 – Radiculalgies et syndromes canalaires
| Rang | Rubrique | Intitulé | Descriptif |
|---|---|---|---|
 |
Définition | Connaître la définition des termes de syndrome canalaire, radiculopathie et plexopathie | |
|
Diagnostic positif | Savoir diagnostiquer une radiculalgie et ses formes topographiques | |
|
Diagnostic positif | Savoir évoquer le diagnostic de plexopathie | Caractéristiques sémiologiques du syndrome de Pancoast-Tobias (plexopathie infiltrative) |
|
Étiologie | Connaître les étiologies des radiculalgies | |
 |
Diagnostic positif | Connaître les diagnostics différentiels des radiculalgies | |
|
Identifier une urgence | Identifier les situations d’urgence | |
|
Examens complémentaires | Connaître l’indication et la hiérarchisation des demandes d’examens complémentaires devant une radiculalgie ou plexopathie | |
|
Prise en charge | Argumenter le traitement de première intention d’une radiculalgie non compliquée | Traitement symptomatique : traitement médicamenteux et non médicamenteux |
|
Éléments physiopathologiques | Connaître la physiopathologie des syndromes canalaires | |
|
Diagnostic positif | Savoir faire le diagnostic d’un syndrome canalaire | Caractéristiques sémiologiques d’un syndrome du canal carpien, d’une atteinte du nerf ulnaire au coude |
|
Examens complémentaires | Indication et hiérarchisation des demandes d’examens complémentaires devant un syndrome canalaire | |
|
Identifier une urgence | Savoir reconnaître les signes de gravité d’un syndrome canalaire | |
|
Diagnostic positif | Connaître les diagnostics différentiels d’un syndrome canalaire | |
|
Étiologies | Connaître les étiologies des syndromes canalaires | |
|
Diagnostic positif | Connaître les différentes formes topographiques les plus fréquentes des syndromes canalaires et radiculaires | Syndrome du canal carpien, syndrome de compression du nerf ulnaire au coude |
|
Diagnostic positif | Connaître les autres formes topographiques des syndromes canalaires | Compression du nerf fibulaire au col de la fibule, atteinte du nerf radial dans la gouttière humérale |
|
Prise en charge | Argumenter le traitement de première intention d’un syndrome canalaire non compliqué | |
|
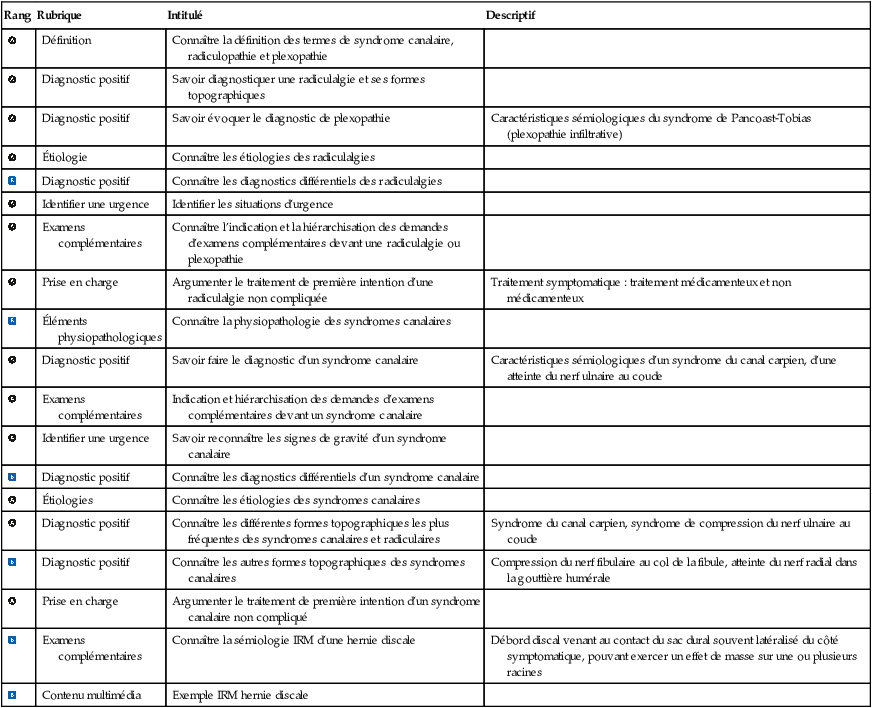
Examens complémentaires | Connaître la sémiologie IRM d’une hernie discale | Débord discal venant au contact du sac dural souvent latéralisé du côté symptomatique, pouvant exercer un effet de masse sur une ou plusieurs racines |
|
Contenu multimédia | Exemple IRM hernie discale |
106Item 96 – Neuropathies périphériques
| Rang | Rubrique | Intitulé | Descriptif |
|---|---|---|---|
|
Définition | Connaître la classification et la définition des différents types de neuropathies périphériques | Mononeuropathie, mononeuropathie multiple, plexopathie, polyneuropathie, polyradiculoneuropathie, neuronopathie |
|
Diagnostic positif (examen clinique, démarche diagnostique) | Connaître les modalités du diagnostic d’une neuropathie périphérique | Appliquer une démarche de localisation, mécanisme physiopathologique, classification électro-clinique |
|
Diagnostic positif (examen clinique, démarche diagnostique) | Connaître les modalités du diagnostic d’une polyneuropathie, d’une mononeuropathie, d’une mononeuropathie multiple, d’une polyradiculoneuropathie | |
|
Diagnostic positif (examen clinique, démarche diagnostique) | Connaître les modalités du diagnostic d’une neuronopathie sensitive | |
|
Diagnostic positif | Distinguer cliniquement neuropathie périphérique et sclérose latérale amyotrophique | Atteinte motrice pure, anomalies centrales et périphériques |
|
Étiologies | Connaître les étiologies des principales polyneuropathies axonales longueur-dépendantes | Polyneuropathies des diabétiques, toxiques (médicamenteuses) et éthyliques |
|
Étiologies | Connaître les étiologies des polyneuropathies axonales longueur-dépendantes plus rares | Amylose |
|
Étiologies | Connaître les étiologies des principales polyneuropathies démyélinisantes | IgM à activité anti-MAG, maladie de Charcot-Marie-Tooth |
|
Étiologies | Connaître les étiologies des principales neuronopathies | Neuronopathies paranéoplasiques |
|
Étiologies | Connaître les étiologies des principales mononeuronopathies multiples | Vascularite du nerf |
|
Identifier une urgence | Connaître l’urgence du diagnostic et de la prise en charge des mononeuropathies multiples | |
|
Examens complémentaires | Avoir une stratégie d’utilisation des examens complémentaires | Explorations biologique, neurophysiologique, liquide cérébrospinal, biopsie neuromusculaire |
107Item 97 – Polyradiculonévrite aiguë inflammatoire (syndrome de Guillain-Barré)
| Rang | Rubrique | Intitulé | Descriptif |
|---|---|---|---|
|
Définition | Connaître la définition d’une polyradiculonévrite | |
|
Définition | Connaître les différentes formes de polyradiculonévrites | Syndrome de Guillain-Barré, neuropathie motrice axonale aiguë |
|
Diagnostic positif | Connaître les modalités du syndrome de Guillain-Barré | Savoir évoquer le diagnostic de syndrome de Guillain-Barré devant l’apparition d’une parésie symétrique, étendue et sévère, prédominant initialement en proximal |
|
Étiologies | Connaître l’existence d’événements déclenchant des polyradiculonévrites | |
|
Diagnostic positif | Reconnaître les trois phases d’évolution d’une polyradiculonévrite | |
|
Examens complémentaires | Connaître les modalités d’une ponction lombaire évocatrice d’un syndrome de Guillain-Barré | Savoir interpréter un, liquide cérébrospinal de syndrome de Guillain-Barré (hyperprotéinorachie marquée pauci- ou acellulaire) |
|
Identifier une urgence | Connaître les signes de gravité d’une polyradiculonévrite aiguë et les situations nécessitant une prise en charge en service de réanimation | Connaître les risques respiratoire, thromboembolique et cardiaque, savoir rechercher une paralysie faciale bilatérale, rechercher les troubles de la déglutition et de la phonation |
|
Prise en charge | Connaître les principes de prise en charge d’un syndrome de Guillain-Barré |
108 L’atteinte du système nerveux périphérique est définie par l’ensemble des manifestations cliniques, électriques, biologiques et histologiques résultant d’une atteinte du neurone périphérique.
Le système nerveux périphérique est composé de (fig. 8.1 et 8.2) :
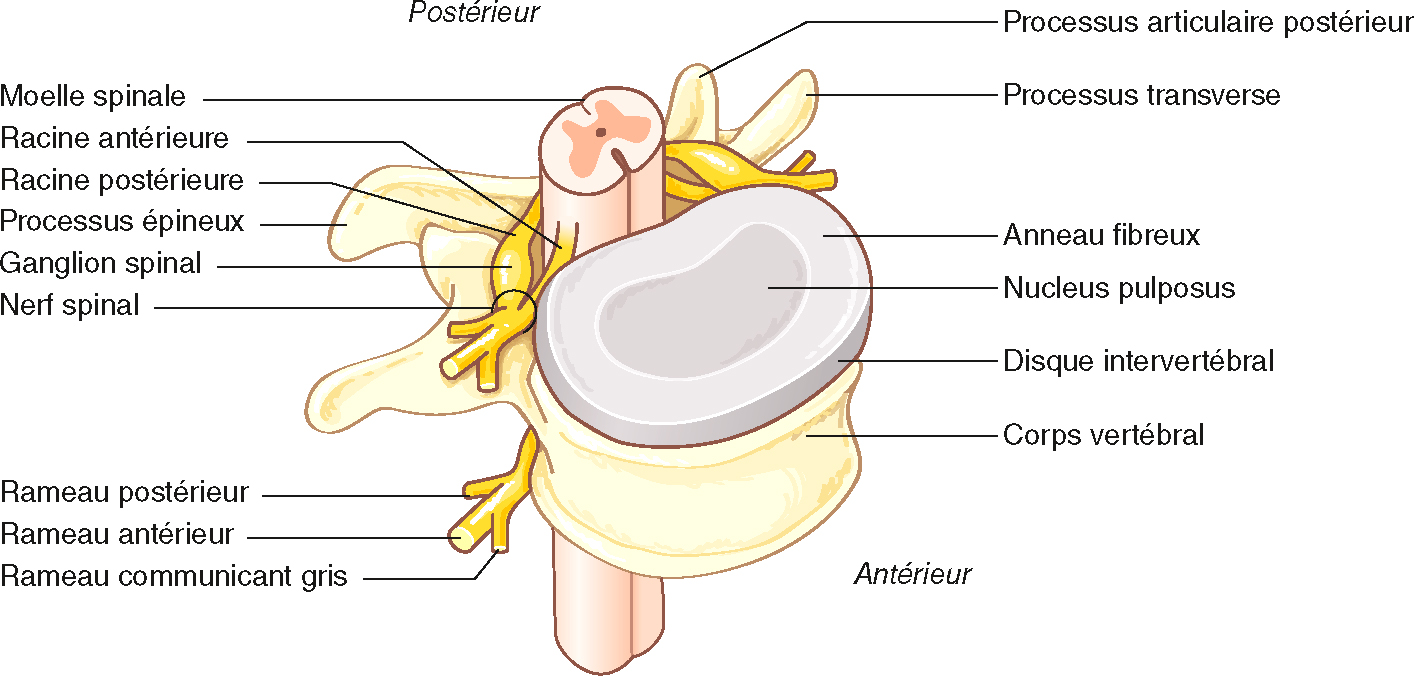
Système nerveux périphérique : émergence du nerf spinal de la moelle spinale. Illustration de Carole Fumat.
Diagramme anatomique montrant le développement nerveux dans les nerfs de la colonne vertébrale de la colonne vertébrale. La forme (sensibilité) de la racine antérieure (moteur) et les racines dorsales se forme, et à l'approche de la colonne vertébrale, les nerfs des vertèbres proviennent du contremaître des vertèbres se forment. Afin de se connecter au système nerveux autonome, les nerfs sont ensuite divisés en branches antérieures et postérieures et déterminées, telles que les branches de communication grise. La photographie met également en évidence la structure du volet, composée de la marque centrale de la colonne vertébrale, des processus articulaires épineux, latéraux et postérieurs, et des fibres externes et du noyau vertébral central. Ce diagramme montre le tissu fonctionnel et mécanique entre le système nerveux périphérique et la colonne vertébrale.
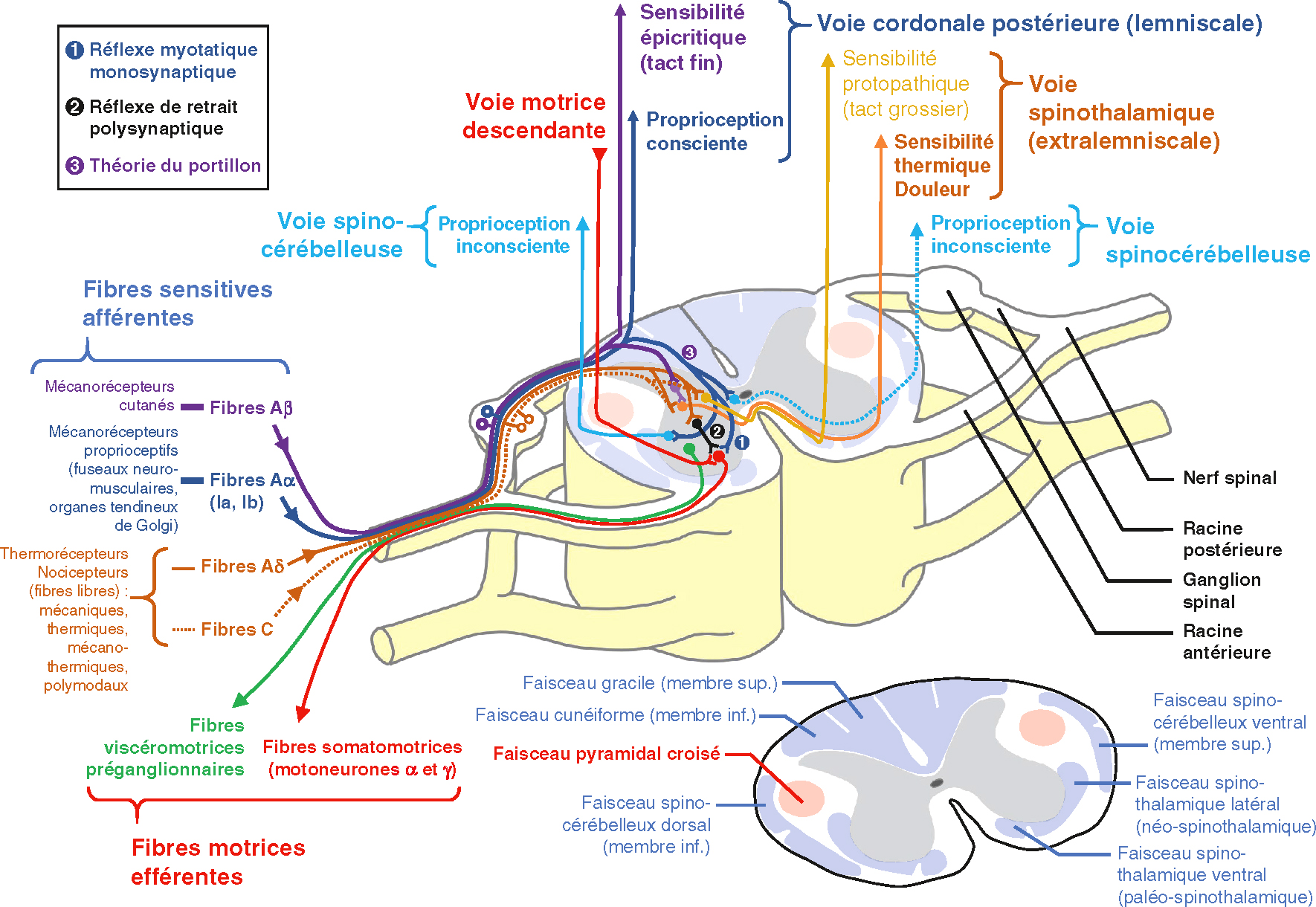
Système nerveux périphérique : fibres sensitives, nerf spinal et voies ascendantes.Des diagrammes anatomiques et fonctionnels détaillés du système nerveux périphérique montrant des espèces sensorielles pertinentes, des fibres motrices efférentes et les voies ascendantes les plus importantes de la moelle épinière. Sur le côté gauche se trouve une variété de fibres sensibles (Aβ, Aδ, C, Ia / Ib) de la peau, avec des récepteurs proprioceptifs et nociceptifs qui traversent les racines dorsales pour atteindre la colonne vertébrale. Les fibres végiomotrices préganglionnaires et somatomotrices efférentes partent de la racine antérieure. Le trajet des voies ascendantes est codé en couleurs : la voie lemniscale (en bleu) transmet la proprioception consciente et le tact fin, tandis que la voie spinothalamique (en orange) relaie la douleur et la sensibilité thermique. La voie spinocérébelleuse (en turquoise) transmet la proprioception inconsciente. Le faisceau pyramidal croisé (en rouge) illustre la voie motrice descendante. Des circuits réflexes (myotatique monosynaptique et de retrait polysynaptique) complètent l’organisation intégrée des réponses motrices.
- • fibres motrices efférentes : le corps cellulaire est situé dans la corne antérieure de la moelle spinale; l’axone quitte la moelle spinale par la racine antérieure et chemine jusqu’au muscle strié squelettique où il tient sous sa dépendance une centaine de fibres (unité motrice);
-
• fibres sensitives afférentes : le corps cellulaire est situé dans le ganglion spinal de la racine postérieure; leur destinée médullaire les oppose en trois groupes :
- – lemniscales, dont les fibres cheminent dans les cordons postérieurs homolatéraux et véhiculent la sensibilité épicritique et proprioceptive consciente;
- – spinothalamiques, dont les fibres, qui décussent au niveau du métamère médullaire, véhiculent de façon controlatérale le tact grossier et la thermoalgie;
- – spinocérébelleuses, dont les fibres décussent au niveau du métamère médullaire pour le membre supérieur et empruntent une voie directe pour le membre inférieur, véhiculant la proprioception inconsciente;
- • fibres végétatives (voies efférentes sympathiques et parasympathiques) : le corps cellulaire du neurone préganglionnaire est situé dans le tronc cérébral ou la moelle spinale; l’axone quitte le système nerveux central par le trajet des nerfs crâniens (III, VII, IX, X) – uniquement pour le contingent parasympathique – ou des racines antérieures médullaires pour faire un relais avec le neurone post-ganglionnaire innervant muscles lisses et glandes.
Les fibres nerveuses sont constituées d’axones (cellules nerveuses), de cellules de Schwann (myéline) et de tissus de soutien (vaisseaux et conjonctif). Les lésions élémentaires des fibres nerveuses sont :
- • la dégénérescence wallérienne : désintégration progressive myélino-axonale, puis bouquets de régénérescence axonale (clusters);
- • la démyélinisation segmentaire : mise à nu progressive des axones par destruction de la myéline au niveau des nœuds de Ranvier qui s’élargissent anormalement.
109Au plan anatomique, le système nerveux périphérique débute à l’émergence des radicelles ventrales et dorsales à partir de la moelle épinière. Ces radicelles fusionnent en racines ventrales et dorsales. Ces paires de racines se rejoignent pour former un nerf spinal (souvent appelé à tort « racine nerveuse ») au trou de conjugaison intervertébral. Le nerf spinal quitte le canal vertébral à travers le foramen intervertébral. Ce nerf est court et se divise presque immédiatement en un rameau dorsal et ventral. Les rameaux ventraux cervicaux, lombaires et sacrés vont former des plexus. Par exemple, le plexus cervical est formé de l’union des rameaux ventraux des nerfs spinaux (appelés à tort « racines ») C5 à D1. Les interconnexions qui s’effectuent dans le plexus résultent en la formation de trois troncs nerveux importants (radial, médian, ulnaire) et d’autres nerfs moins importants au membre supérieur (fig. 8.3).
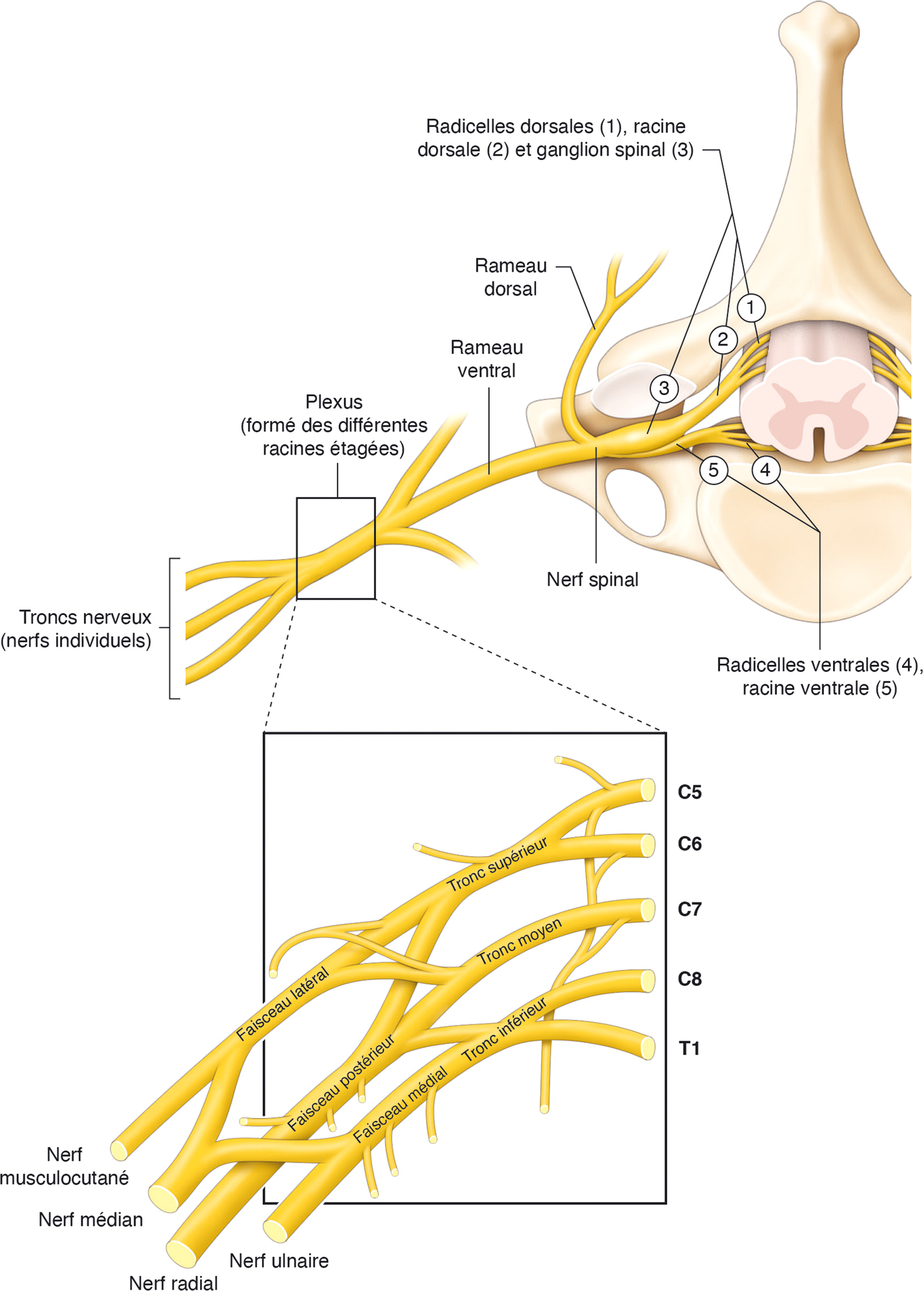
Organisation anatomique de l’émergence des radicelles, des racines nerveuses et du plexus brachial. Illustration de Cyrille Martinet.
Diagrammes anatomiques décrivant les maladies neurologiques, le développement des racines vertébrales, l'organisation de la formation du plexus brachial. À droite, sur le côté droit, la moelle épinière est indiquée dans la section transversale, montrant les frais de licence dorsale (1), la racine dorsale (2), le ganglion vertébral (3), les ragins abdominaux (4) et la racine abdominale (5). Ceci est divisé en branches dorsales et ventrales. Les branches abdominales contribuent à la composition du plexus brachial supérieur. Le plexus brachial est montré en bas avec des articles dans divers segments de C5 à T1. Les troncs supérieurs, moyens et inférieurs s'étendent dans les faisceaux latéraux, postérieurs et médiaux, conduisant aux nerfs périphériques les plus importants, aux cacans musculaires, médian, radial et ulna.
Item 95 – Radiculalgies et syndromes canalaires
- I 110111Mononeuropathies : syndromes canalaires et compressions posturales
- II Radiculopathies
- III Atteintes plexiques (plexopathies)
I Mononeuropathies : syndromes canalaires et compressions posturales
- • Les syndromes canalaires sont liés à des compressions des nerfs périphériques dans des zones anatomiques étroites pour le cheminement du nerf.
- • Les compressions dites posturales surviennent dans des zones anatomiques localisées où le nerf est superficiel et donc vulnérable à une compression externe.
-
•
La symptomatologie est importante à connaître pour établir le diagnostic différentiel avec les radiculalgies (tableau 8.1, fig. 8.4 et 8.5). Synthèse des territoires tronculaires et radiculaires.Racines à l’origine des troncs nerveux Tronc nerveux Principaux muscles innervés Réflexe Membre supérieur C6
C7
C8Médian Opposant
Court abducteur du poucePalmaire C6
C7Radial Extenseurs des doigts Stylo-radial C8-T1 Ulnaire Interosseux Ulno-pronateur Membre inférieur L3-L4 Fémoral Quadriceps Patellaire (rotulien) L4-L5
L5Nerf fibulaire commun Releveurs du pied
Extenseur du 1er orteil– S1 Nerf tibial Extenseurs du pied (gastrocnémien et soléaire)
Interosseux
Fléchisseurs des orteilsCalcanéen (achilléen) 
Source : CEN, 2018.
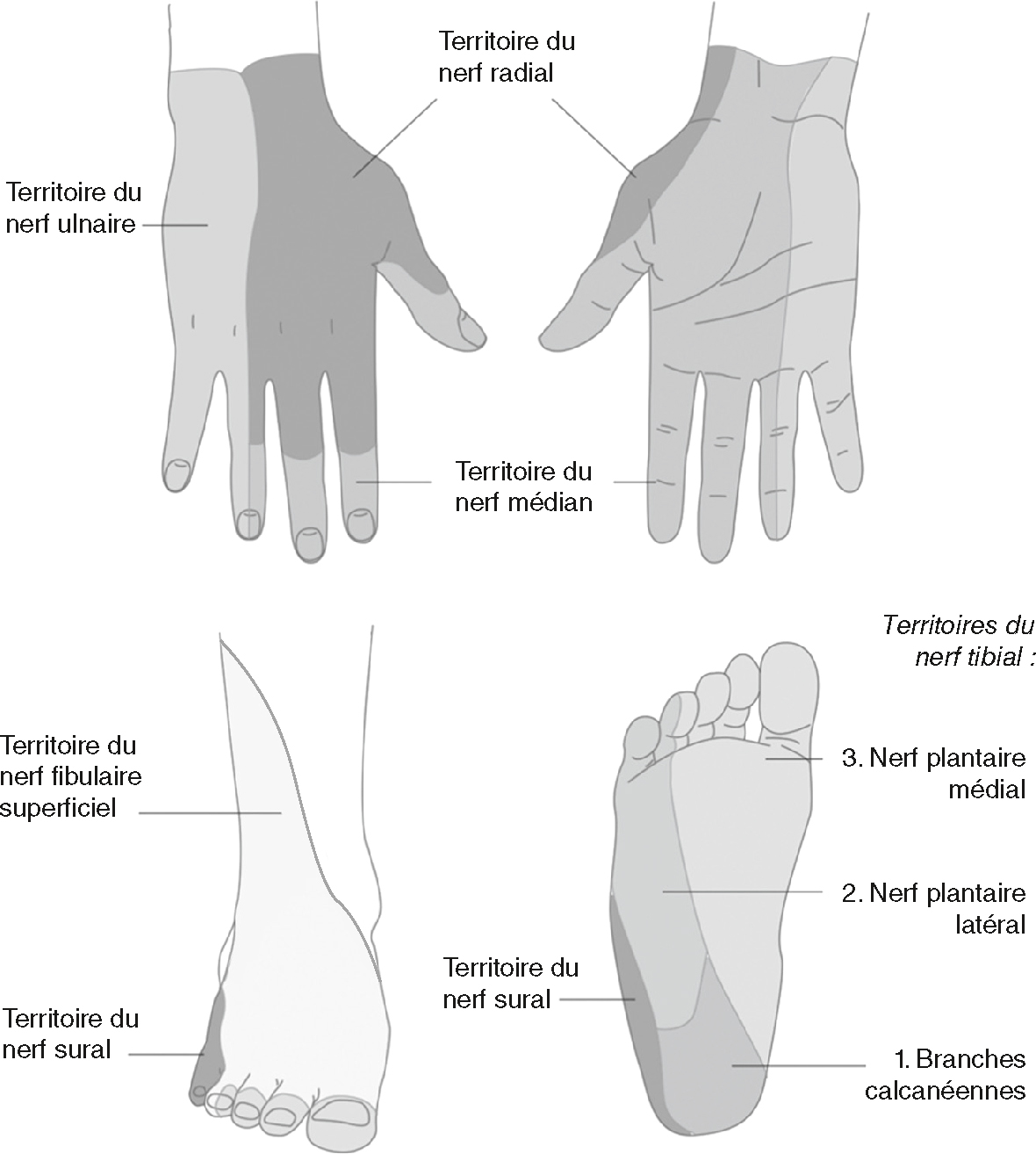
Fig. 8.4 Territoires tronculaires.
Illustration d’Hélène Fournié.Diagrammes anatomiques décrivant les maladies neurologiques, le développement des racines vertébrales, l'organisation de la formation du plexus brachial. À droite, sur le côté droit, la moelle épinière est indiquée dans la section transversale, montrant les frais de licence dorsale (1), la racine dorsale (2), le ganglion vertébral (3), les ragins abdominaux (4) et la racine abdominale (5). Ceci est divisé en branches dorsales et ventrales. Les branches abdominales contribuent à la composition du plexus brachial supérieur. Le plexus brachial est montré en bas avec des articles dans divers segments de C5 à T1. Les troncs supérieurs, moyens et inférieurs s'étendent dans les faisceaux latéraux, postérieurs et médiaux, conduisant aux nerfs périphériques les plus importants, aux cacans musculaires, médian, radial et ulna.

Fig. 8.5 Territoires radiculaires.
Illustration d’Hélène Fournie.Vues anatomiques de la vue avant supérieure et de la vue arrière du membre inférieur.Cela montre la région neurobulaire sensible à la peau correspondant aux racines du nerf vertébral. Chaque zone est caractérisée par des ligaments gris de force différemment, avec des racines C6, C7 et C8 des racines cutanées représentant des dermatomes innocents dans les bras, et le bas du dos des pieds et des racines sacrées L4, L5 et S1. Dans les membres supérieurs, c'est la surface extérieure du pouce de l'avant-bras, avec C7 s'étendant à l'arrière de l'avant-bras jusqu'au majeur, et C8 s'étendant à l'oreille du côté médial de l'avant-bras. À des niveaux inférieurs, L4 couvre la face avant et intérieure des cuisses, les jambes couvrant les marleurelas intérieures, L5 suit le côté extérieur de la cuisse, les pattes avant continuant vers le grand orteil. Cette carte dermatologique vous permet de relier les zones du corps aux racines nerveuses exactes de la douleur, des hypochondries ou du parathyme.
- • 112 Le trouble initial est lié à un blocage de la propagation de l’influx secondaire à la compression (bloc de conduction). Lorsque la compression est sévère et prolongée, les lésions initiales se compliquent d’une dégénérescence axonale.
-
• L’ENMG montre le bloc de conduction et établira l’importance de la perte axonale éventuelle (voir fig. 8.12); schématiquement :
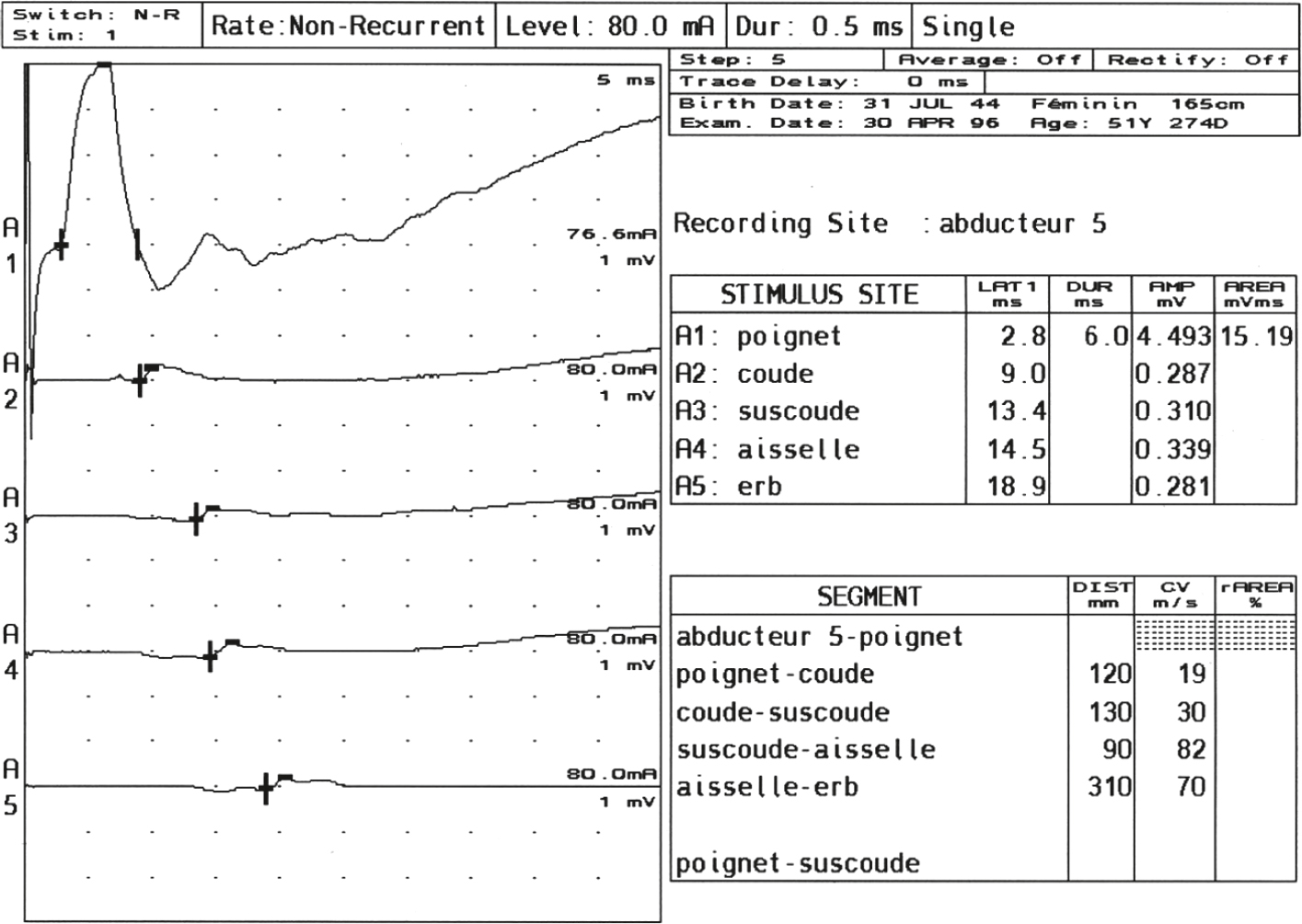
Fig. 8.12 Aspect de bloc de conduction (vitesse de conduction motrice) proximal du nerf ulnaire dans une neuropathie motrice multifocale.On observe une réponse normale au poignet avec une amplitude correcte (15.19 mV·ms), mais une chute brutale d’amplitude à partir du coude (0.287 mV·ms) et au-delà. Le tableau indique une vitesse de conduction très ralentie entre le coude et la sus-coude (19 m/s), traduisant un bloc de conduction proximal. Cet aspect électrophysiologique est évocateur d’une neuropathie motrice multifocale, caractérisée par des blocs focaux segmentaires sur les nerfs moteurs sans atteinte sensitive.
- – l’atteinte initiale se traduit par un ralentissement ou un blocage de la conduction nerveuse;
-
– la dégénérescence axonale se traduit par une diminution de la réponse motrice ou sensitive en aval du site lésionnel avec des signes de dénervation des fibres musculaires (tracé neurogène) (voir fig. 8.10).
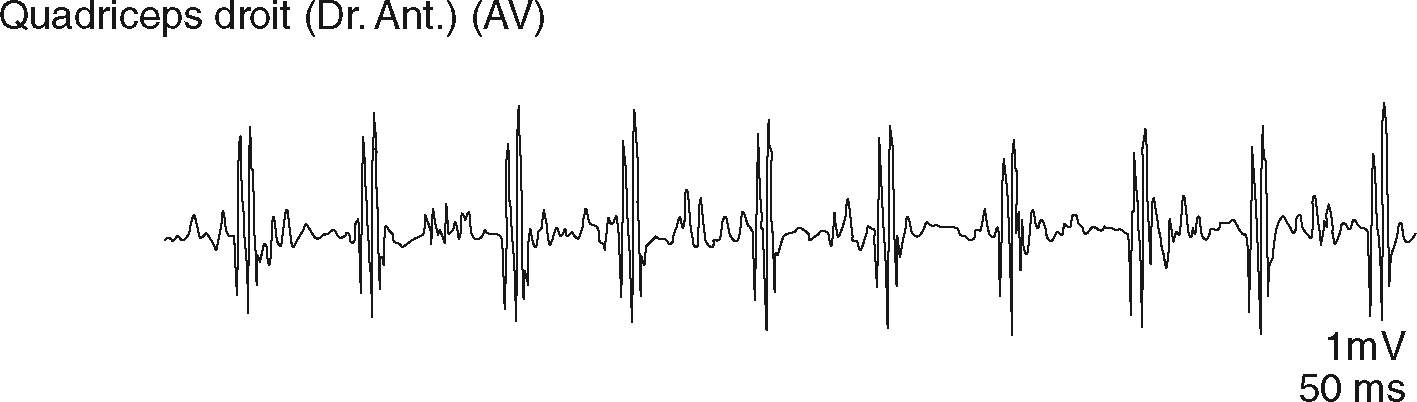
Fig. 8.10 Tracé neurogène de dénervation chronique : potentiel polyphasique se répétant à une fréquence élevée (sommation temporelle).L’enregistrement électromyographique du muscle quadriceps droit montre une activité typique d’une dénervation chronique avec réinnervation. Le tracé met en évidence des potentiels d’unité motrice polyphasiques, de forte amplitude, se répétant à intervalles réguliers et à fréquence élevée, illustrant une nette sommation temporelle. La ligne de base reste relativement stable entre les salves, ce qui accentue la lisibilité des réponses musculaires. Ce type de signal est généralement observé dans les atteintes neurogènes où les motoneurones restants recrutent un plus grand nombre de fibres musculaires, traduisant un processus adaptatif. L’amplitude atteint ici 1 millivolt et chaque division horizontale correspond à 50 millisecondes, ce qui permet d’apprécier la densité temporelle des potentiels. L’aspect polyphasique et répétitif des unités motrices est un signe classique d’une réorganisation musculaire secondaire à une perte initiale d’innervation. Le tracé met ainsi en évidence une activité neuromusculaire compensatoire, révélatrice d’un processus de réinnervation fonctionnelle.
A 113Clinique
- • Le plus souvent, il s’agit d’une atteinte de troncs nerveux sensitivo-moteurs (nerfs mixtes).
- • Plus rarement, la symptomatologie est purement sensitive ou à prédominance sensitive (paresthésies).
- • 114Une atteinte purement motrice doit conduire à remettre le diagnostic en cause (voir plus loin Item 96 – Neuropathies périphériques).
- • L’examen objective le territoire anatomique du déficit moteur et sensitif.
-
• Une amyotrophie traduit l’existence d’une atteinte axonale déjà évoluée (fig. 8.6).
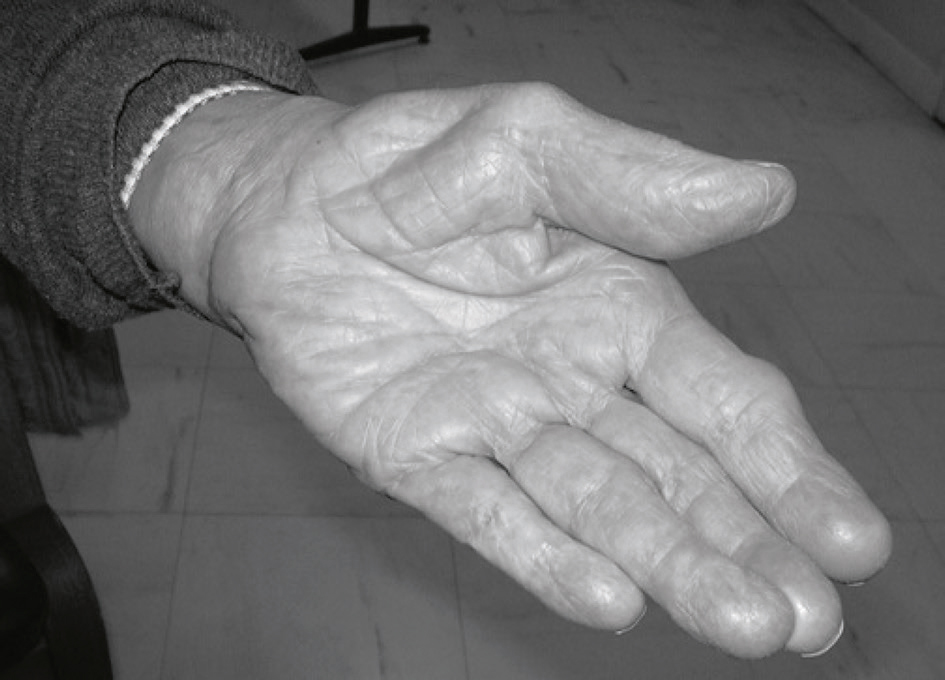
Fig. 8.6 Amyotrophie sévère du muscle court abducteur du pouce en cas de syndrome du canal carpien évolué.Main gauche d’un patient en légère supination avec les doigts relâchés, laissant apparaître une dépression marquée dans la région thénarienne. Cette cavité bien visible à la base du pouce est due à une amyotrophie sévère du muscle court abducteur, caractéristique d’un syndrome du canal carpien évolué. L’affaiblissement musculaire est localisé, sans altération visible des autres éminences de la main. Les tendons et plis cutanés sont accentués par la perte de volume, soulignant l’atrophie installée. L’aspect de la peau reste intact, sans rougeur ni œdème, et l’environnement neutre met en valeur les contours anatomiques sans distraction. L’ensemble évoque une atteinte nerveuse chronique liée à la compression prolongée du nerf médian, traduite ici par une perte fonctionnelle manifeste du pouce.
B Examens complémentaires
1 Électroneuromyographie (vidéo 8.1)
L’ENMG est fondamental au double plan diagnostique et pronostique.
-
• L’EMG proprement dit (examen de détection) :
- – précise la topographie tronculaire de l’atteinte motrice;
- – décèle des signes neurogènes dans les muscles atteints;
- – détecte des signes d’activités spontanées de dénervation en faveur d’une dégénérescence axonale;
- – suit la récupération (augmentation de l’amplitude du potentiel moteur, diminution du pourcentage de bloc de conduction, amélioration de la vitesse de conduction).
-
• L’étude de la conduction nerveuse (examen de stimulodétection) évalue sur les fibres motrices :
- – un ralentissement localisé ou bloc de conduction;
- – une diminution d’amplitude du potentiel d’action distal (atteinte axonale et perte en fibres);
- – une diminution d’amplitude du potentiel d’action sensitif.
-
• 115Cette atteinte localisée définit le site de la compression :
- – nerf médian au canal carpien;
- – nerf ulnaire au coude;
- – nerf fibulaire au col du péroné.
- • Une exploration élargie aux autres nerfs permet quelquefois de déceler des atteintes infra-cliniques dans d’autres sites canalaires ou de mettre en évidence une neuropathie diffuse (diabète, neuropathie par susceptibilité à la pression).
2 Autres examens
Le plus souvent, aucun autre examen n’est nécessaire. Un bilan biologique simple peut être effectué, comprenant :
- • glycémie à jeun;
- • numération formule sanguine avec vitesse de sédimentation (augmentée en cas de vascularite).
Et, selon le contexte, toujours dans un second temps :
- • recherche d’une délétion du gène de la PMP22 (neuropathie par hypersensibilité à la pression);
- • recherche d’une hypothyroïdie;
- • recherche d’une mutation du gène de la transthyrétine;
- • recherche de dépôts de substance amyloïde sur une biopsie des glandes salivaires;
- • à part, car contexte évident, hormone chorionique gonadotrophique (β-HCG) au cours de la grossesse.
C Signes de gravité
1 Clinique
- • La présence d’un déficit moteur constitué.
- • L’apparition d’un déficit moteur.
- • Une amyotrophie constituée ou se développant.
- • La douleur en elle-même n’est pas un signe de gravité.
2 Neurophysiologie
- • L’existence de signes d’activités spontanées de dénervation en faveur d’une dégénérescence axonale.
- • La diminution de l’amplitude du potentiel moteur.
D Diagnostics différentiels
- • Une mononeuropathie, forme débutante de mononeuropathie multiple, secondaire à une vascularite. Une mononeuropathie douloureuse aiguë est suspecte de vascu-larite. Un ENMG à la recherche de signes en faveur d’une mononeuropathie multiple est urgent, pour une prise en charge thérapeutique urgente.
- • Une radiculopathie. L’existence d’un trouble sensitif, ou de douleurs dépassant le territoire tronculaire, ou d’un déficit moteur proximal permet d’évoquer le diagnostic. L’ENMG rétablit le diagnostic de radiculopathie en montrant une normalité du potentiel sensitif et des signes neurogènes dans le territoire atteint.
- • 116Une neuropathie motrice à blocs de conduction persistants. L’atteinte est motrice pure et associée à la présence d’anticorps anti-GM1 dans plus de la moitié des cas. Le bloc de conduction se situe en dehors des zones de rétrécissement anatomique (voir plus loin II. Distinguer cliniquement une neuropathie périphérique et une sclérose latérale amyotrophique).
- • Une forme débutante de maladie du motoneurone (sclérose latérale amyotrophique). L’atteinte est motrice pure. La topographie du déficit moteur n’est pas tronculaire (voir plus loin II. Distinguer cliniquement une neuropathie périphérique et une sclérose latérale amyotrophique).
E Grands syndromes
1 Nerf médian au canal carpien (le plus fréquent) (vidéo 8.2)
a Symptômes et signes sensitifs
- • A Paresthésies, souvent douloureuses, nocturnes, systématisées dans le territoire du nerf médian (trois premiers doigts) et affectant préférentiellement le côté dominant.
- • Hypoesthésie à tous les modes de la face palmaire des trois premiers doigts.
- • Fréquent signe de Tinel (dysesthésies dans les doigts lors de la percussion du canal carpien) et un signe de Phalen (reproduction de l’engourdissement et des paresthésies lors de la flexion forcée du poignet).
b Symptômes et signes moteurs
- • Faiblesse de la main, lâchage d’objets, difficulté pour fermer les boutons ou tourner une clé dans une serrure.
- • Déficit des muscles de la partie latérale de l’éminence thénar (court abducteur du pouce, opposant).
- • Tardivement, amyotrophie thénarienne.
c Causes
- • Le plus souvent mécaniques (hyperactivité, travail manuel).
- • Penser aux causes plus rares :
-
- – neuropathie amyloïde (y penser en cas de découverte ou d’une association à une polyneuropathie axonale avec dysautonomie);
- – hypothyroïdie;
- – grossesse.
- –
d Traitement
- • Parfois spontanément résolutif si peu sévère et à prédominance sensitive.
- • Dans un premier temps, infiltration de corticoïdes dans le canal carpien.
- • En cas d’échec : chirurgie (section du ligament transverse du carpe).
2 Nerf ulnaire au coude
a Symptômes et signes sensitifs
- • Paresthésies des 4e et 5e doigts.
- • Hypoesthésie du territoire du nerf à la main (4e et 5e doigts).
b 117Symptômes et signes moteurs
- • Déficit moteur des muscles intrinsèques de la main (interosseux) et respect du muscle fléchisseur ulnaire du carpe.
- • Amyotrophie des espaces interosseux (surtout le 1er espace) et de l’éminence hypothénar.
c Causes
- • Compression dans la gouttière épitrochléo-olécrânienne, notamment lors de l’appui prolongé (ex. : le coude appuyé sur une table).
- • Ou compression par un cal osseux ancien consécutif à une fracture du coude.
d Traitement
- • Immobilisation du coude par attelle de repos dans un premier temps.
- • Traitement chirurgical, si nécessaire (transposition du nerf ulnaire de la gouttière épitrochléo-olécrânienne à la partie antéromédiale de l’avant-bras).
3 Nerf radial dans la gouttière numérale
a Symptômes et signes sensitifs
- • Peu ou pas présents.
- • Territoire : 1er espace interosseux dorsal.
b Symptômes et signes moteurs
- • Déficit moteur de l’ensemble du territoire sous-jacent (muscle brachioradial et extenseurs des doigts et du poignet) responsable d’une main tombante.
- • Respect du muscle triceps et du réflexe tricipital.
c Causes
- • Fractures de l’humérus.
- • Lors de compression externe prolongée du nerf dans la gouttière humérale (paralysie posturale).
d Traitement
La compression externe prolongée est le plus souvent spontanément résolutive, mais la chirurgie peut être nécessaire en cas de fracture humérale.
4 Nerf fibulaire
a Symptômes et signes sensitifs
- • Déficit sensitif discret intéressant une zone cutanée du dos du pied (cou-de-pied).
- • Pouvant s’étendre sur la face latérale de la jambe.
b Symptômes et signes moteurs
- • Déficit des muscles de la loge antérolatérale de jambe (fibulaire, tibial antérieur, long extenseur des orteils, long extenseur de l’hallux, court extenseur des orteils).
- • L’ensemble réalise un steppage lors de la marche.
c 118Causes
- • Compression externe aiguë indolore : paralysie posturale :
-
- – appui prolongé sur la région de la tête de la fibula;
- – position assise jambes croisées;
- – position allongée et amaigrissement (séjour en réanimation);
- – travail en position agenouillée.
- • Vascularite : paralysie aiguë douloureuse.
d Traitement
- • Abstention en cas d’atteinte posturale aiguë.
- • Chirurgie en cas de formes progressives ou si une cause locale de compression a été trouvée (kyste synovial articulaire de la fosse poplitée).
II Radiculopathies
Les radiculopathies sont les pathologies de la racine nerveuse, zone du système nerveux périphérique située entre l’émergence de la moelle épinière et le foramen intervertébral (fig. 8.1).
A Complications neurologiques de la pathologie rachidienne lombaire
Ces complications sont dues le plus souvent à des lésions dégénératives osseuses acquises (arthrosiques) et/ou discales (conflit discoradiculaire), responsables d’une sténose progressive du canal vertébral. Plus rarement, il peut s’agir d’une cause tumorale, telles les lésions intra-durales (tumeurs radiculaires : neurinomes; du filum : épendymomes) et les lésions épidurales (métastases ostéo-épidurales) ou infectieuses (spondylodiscites).
Aux membres inférieurs, les radiculopathies L5 et S1 sont improprement appelées « scia-tiques » . Le nerf sciatique étant le principal tronc nerveux du membre inférieur, son atteinte ne correspond pas à une radiculopathie.
1 Symptômes cliniques
La symptomatologie comporte :
- • des douleurs rachidiennes lombaires pouvant être aiguës (lumbago responsable d’une contracture réflexe), volontiers à l’issue d’un effort de soulèvement, ou chroniques évoluant de façon insidieuse;
- • 119une douleur radiculaire (radiculalgie) :
-
- – unilatérale, partant de la région lombaire basse et adoptant le trajet correspondant à la racine concernée jusqu’au pied :
-
- – L5 : dessus du pied (cou-de-pied) jusqu’au gros orteil,
- – S1 : dessous du pied;
- – ce trajet peut être tronqué, c’est-à-dire limité à une zone du trajet radiculaire complet, par exemple jusqu’au genou ou uniquement à la jambe et au pied; la douleur est typiquement mécanique, accentuée par les efforts, la station debout, calmée par le décubitus; elle peut être impulsive à la toux ou aux efforts de défécation :
-
- – radiculalgie S1 : face postérieure de la fesse, de la cuisse, du mollet jusqu’au talon, plante du pied et 5e orteil,
- – radiculalgie L5 : partie postérolatérale de la cuisse, face latérale de jambe jusqu’au dos du pied et aux premiers orteils; quelquefois trajet inguinal associé,
- – radiculalgie L4 : face antérolatérale de la cuisse, bord antérieur de la jambe, malléole médiale, rarement gros orteil,
- – radiculalgie L3 : partie postéromédiale puis antéromédiale de la cuisse, sans dépasser le genou;
- • un déficit moteur :
-
- – mineur ou modéré, fréquemment observé, correspondant aux muscles innervés par la racine correspondante,
- – complet et aigu en cas de sciatique paralysante.
2 Examen clinique
Il comporte un examen du rachis lombaire et un examen neurologique.
a Signes rachidiens
Ils sont soit spontanés (effacement de la lordose lombaire, inflexion latérale du côté opposé à la douleur), soit responsables d’une limitation des mouvements (flexion antérieure du tronc : mesure de la distance doigts–sol ou de l’indice de Schöber; inflexions latérales).
b Signes radiculaires
- • Signe de Lasègue : en décubitus dorsal, l’élévation du membre inférieur va reproduire la douleur radiculaire L4, L5 ou S1 à partir d’un certain angle par rapport au plan du lit.
- • Signe de Léri : en décubitus ventral, l’hyperextension de la cuisse sur le bassin, membre inférieur en extension, peut reproduire une douleur L3 ou L4.
- • Signes moteurs déficitaires du territoire radiculaire correspondant.
- • Troubles de la sensibilité dans le territoire radiculaire.
- • Anomalie des réflexes tendineux (voir tableau 8.1) :
-
- – abolition du réflexe calcanéen (achilléen) signant l’atteinte radiculaire S1;
- – abolition du réflexe patellaire (rotulien) lors des atteintes L3 ou L4;
- – à noter : pas de perte de réflexe en cas d’atteinte L5.
120
c Atteinte pluriradiculaire par atteinte de la queue de cheval
- • Interrogatoire : troubles urinaires (perte ou rétention d’urines), anaux (constipation, perte des selles) et sexuels (troubles de l’érection).
- • Examen : insensibilité périnéale.
- • Une compression pluriradiculaire (par exemple, par une hernie discale) doit être recherchée en urgence.
3 Examens complémentaires
- • Pas d’exploration systématique en raison de l’extrême fréquence de la lombalgie et des radiculopathies, en particulier aux membres inférieurs (« sciatique »).
-
• Un bilan complémentaire est demandé devant une aggravation des symptômes et des signes cliniques ou leur persistance (fig. 8.7 et 8.8).
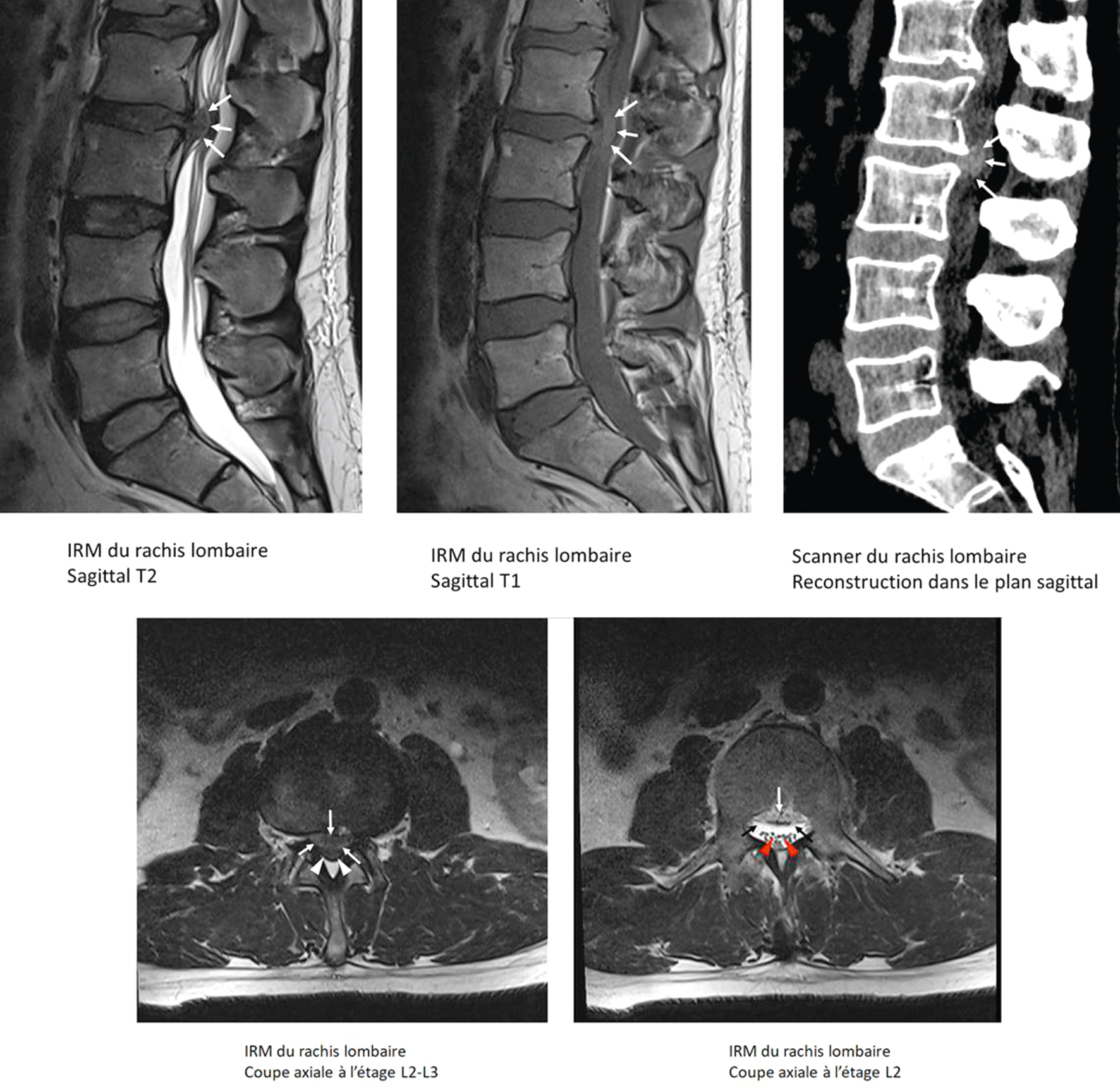
Fig. 8.7 Hernie discale L2-L3 avec protrusion médiane ascendante réalisant une compression focale des racines de la queue de cheval à cet étage.
Coupes sagittales : délimitation de la hernie par les flèches blanches. Coupes transversales : T1 : hernie médiane (flèches blanches) et fourreau dural repoussé (têtes de flèches blanches). T2 : hernie médiane (flèches noires) et fourreau dural repoussé (têtes de flèches rouges).En sagittal T2 (haut gauche), on observe une protrusion discale médiane ascendante avec déformation du sac dural. L’image T1 (haut centre gauche) confirme la perte de signal du disque, traduisant une dégénérescence. Le scanner (haut droite) met en évidence la protrusion calcifiée dans le canal rachidien. Les coupes axiales T2 (bas) à l’étage L2-L3 montrent une compression centrale du contenu canalaire, et l’image inférieure droite avec flèches rouges illustre la compression focale des racines de la queue de cheval. L’aspect global est celui d’une hernie discale comprimant directement les structures nerveuses centrales à ce niveau.
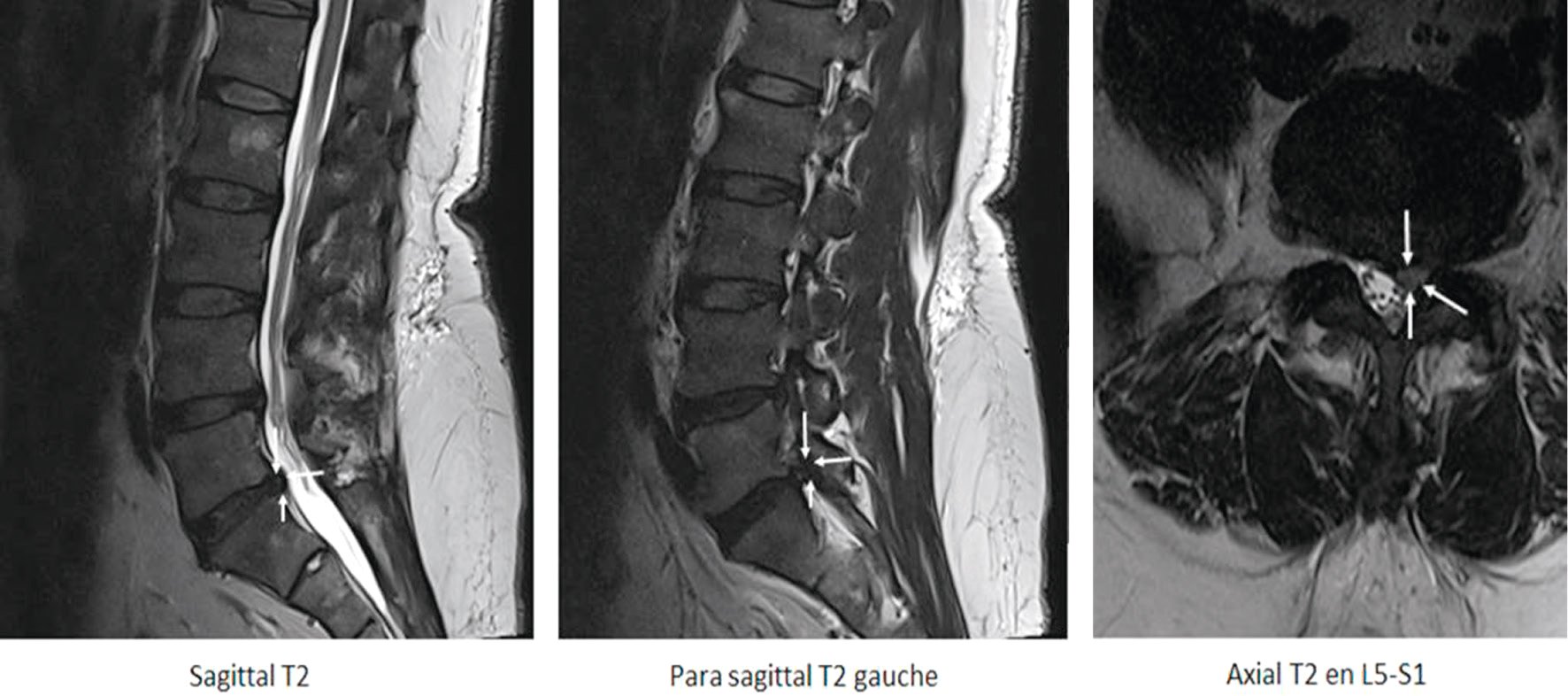
Fig. 8.8 Hernie postérolatérale gauche L5-S1 en hypersignal modéré (potentiellement exclue), conflictuelle avec l’émergence durale de la racine S1 gauche (flèches blanches).Les images en pondération T2 montrent une hernie discale postéro-latérale gauche au niveau L5-S1. Sur la coupe sagittale, on observe une saillie discale en hypersignal modéré qui modifie le contour postérieur du disque, laissant supposer un fragment possiblement exclu. L’image para-sagittale gauche met en évidence l’extension de cette hernie vers le foramen, dans le prolongement du trajet de la racine S1. Sur la coupe axiale, la hernie entre clairement en conflit avec l’émergence durale de la racine S1 gauche, comme l’indiquent les flèches blanches, confirmant l’origine du conflit disco-radiculaire. Ce contact étroit entre la racine et le fragment discal hernié est responsable d’une compression foraminale unilatérale gauche, expliquant les symptômes d’irradiation douloureuse ou de déficit sensitivo-moteur en territoire S1. L’aspect global évoque une hernie à composante extrudée, localisée à gauche, avec effet compressif direct sur la racine adjacente.
- • IRM lombaire :
-
- – en cas de persistance des symptômes, malgré un traitement médical bien conduit au-delà de 6 semaines et/ou d’un déficit neurologique franc;
- – l’IRM permet de confirmer le diagnostic de radiculopathie liée à une pathologie discale ou rachidienne dégénérative, mais elle a également pour objectif de visualiser l’ensemble de la queue de cheval à la recherche d’autres causes plus rares de radiculopathies :
-
- – les lésions intradurales (tumeurs radiculaires : neurinomes; du filum : épendymomes) et les lésions épidurales (métastases ostéo-épidurales),
- – un débord discal venant au contact du sac dural souvent latéralisé du côté sympto-matique responsable d’un conflit (effet de masse) avec la racine passant en regard du disque (par exemple, hernie discale postérolatérale L5 en regard du disque L4-L5), voire plusieurs racines,
- – une hernie foraminale qui comprime la racine sus-jacente (L4 pour le disque L4-L5).
- • Scanner lombaire, moins fréquent, sans et avec injection de produit de contraste :
-
- – les fenêtres osseuses montrent l’arthrose des apophyses articulaires postérieures et le retentissement sur la partie latérale du canal rachidien (canal déformé « en feuille de trèfle »);
- – il montre également la hernie discale et ses rapports avec le canal vertébral.
4 121Traitement
Traitement médical dès le début des symptômes sur les seuls éléments cliniques. L’indication d’un repos doit être minimalisée.
- • Les myorelaxants, antalgiques et anti-inflammatoires sont préconisés.
- • Le traitement sera poursuivi pendant 2 à 4 semaines.
- • Reprise progressive d’activités sous couvert éventuel d’une kinésithérapie appropriée : renforcement des muscles paravertébraux et abdominaux; apprentissage des règles d’hygiène vertébrale.
- • 122En cas d’échec, une prise en charge rhumatologique avec réalisation d’infiltrations peut encore améliorer certains patients.
- • Indication opératoire à discuter en cas d’échec : exérèse de la hernie et curetage discal sous anesthésie générale, laminectomie étendue en cas de sténose canalaire, décompression foraminale, voire stabilisation par arthrodèse et ostéosynthèse en cas de spondylolisthésis.
- • Exploration et indication opératoire urgentes en cas de sciatique paralysante.
B Complications neurologiques de la pathologie rachidienne cervicale
Les manifestations sont consécutives à une hernie discale, apparue le plus souvent sans cause déclenchante, ou aux lésions uncodiscarthrosiques des vertèbres cervicales rétrécissant le foramen vertébral.
1 Symptômes et examen clinique
- • Symptômes : douleur exacerbée par les mouvements du rachis, les efforts de toux et parfois le décubitus.
- • Examen clinique :
-
- – limitation douloureuse des mouvements du rachis cervical;
- – contracture des muscles cervicaux;
- – la mobilisation peut réveiller la douleur radiculaire.
- • Recherche d’un déficit moteur, sensitif, d’une modification (diminution ou abolition) d’un réflexe tendineux.
- • Atteinte radiculaire (névralgie cervicobrachiale) : la cervicalgie intéresse la nuque, la douleur se poursuit sur l’épaule puis le membre supérieur en suivant une topographie orientant vers le niveau radiculaire :
-
- – douleur C5 : face latérale du moignon de l’épaule et du bras;
- – 123douleur C6 : antérieure à l’épaule, au bras, au coude, à l’avant-bras pour se terminer dans les deux premiers doigts de la main (pouce);
- – douleur C7 : postérieure au bras, au coude, à l’avant-bras et à la main dans les trois doigts moyens (index-médius);
- – douleur C8 : bord médial du membre supérieur, se terminant dans les deux derniers doigts;
- – douleur T1 : face médiale du bras.
- • Myélopathie cervicale : il convient de dépister une souffrance médullaire éventuellement associée (atteinte pyramidale, troubles sensitifs et sphinctériens, signe de Lhermitte).
2 Examens complémentaires
L’IRM est l’examen essentiel. Elle objective l’ensemble du rachis cervical et définit le diamètre sagittal du canal rachidien. Elle apprécie l’effacement des espaces sous-arachnoïdiens et d’éventuelles anomalies du signal médullaire sur les séquences pondérées en T2, témoignant d’une souffrance médullaire.
3 Traitement
a Traitement médical
Le traitement médical (repos, anti-inflammatoires, antalgiques, voire corticothérapie par voie orale à raison de 1 mg/kg durant 10 à 15 jours, collier cervical) est toujours préconisé en première intention.
La plupart des névralgies cervicobrachiales guérissent en 4 à 6 semaines.
b Traitement chirurgical
Le traitement chirurgical, d’indication rare, consiste en l’exérèse de la hernie discale (hernie molle généralement post-traumatique) ou, plus rarement, en une décompression de la racine dans le foramen rétréci par un ostéophyte de l’uncus (voie antérieure, un ou plusieurs niveaux selon les données de la clinique et de l’imagerie).
Ce traitement est parfois indiqué en urgence, en cas d’atteinte déficitaire sévère.
En cas d’atteinte médullaire associée, la chirurgie rapide permet une stabilisation des troubles neurologiques anciens et, parfois, des améliorations des déficits récents.
C 124Diagnostic différentiel
- • Une mononeuropathie – syndrome canalaire – lorsque la radiculopathie est tronquée. La douleur est alors limitée à une partie du territoire de la radiculopathie. Par exemple, douleur de la face postérieure de la cuisse au cours d’une radiculopathie S1, douleur du cou-de-pied et de l’espace entre le 1er et le 2e orteil dans une radiculopathie L5.
- • Une méningoradiculite, infectieuse ou infiltrative (voir ci-après).
- • Une plexopathie si les douleurs et le déficit est pluriradiculaire (voir ci-après).
D Méningoradiculites après morsure de tique
Borrelia burgdorferi, spirochète vecteur de la maladie de Lyme transmis par une tique (Ixodes), peut être responsable de manifestations dermatologiques (l’erythema chronicum migrans) à la phase initiale de l’infection.
L’absence de traitement par bêtalactamines ou cyclines peut être suivie :
- • de lésions viscérales polymorphes quelques mois après l’infestation initiale : arthrite, récidive d’erythema chronicum migrans, voire bloc auriculoventriculaire ;
- • et d’une méningoradiculite :
-
- – définie par l’association d’une méningite et d’une atteinte radiculaire,
- – touchant 10 à 20 % des patients.
1 Clinique
Elle est caractérisée par :
- • des douleurs radiculaires très intenses, diurnes et nocturnes, uni- ou pluriradiculaires et asymétriques, associées à un déficit sensitivo-moteur dans les territoires atteints, à proximité du site de la morsure;
- • une atteinte faciale, parfois bilatérale, dans près de 50 % des cas (surtout chez l’enfant). Le syndrome méningé est souvent au second plan et se traduit avant tout par des céphalées.
2 Explorations
- • L’examen du LCS montre une augmentation du nombre de cellules à prédominance lympho-cytaire, une hyperprotéinorachie. L’isoélectrofocalisation des protéines du LCS peut montrer une répartition oligoclonale des protéines.
- • L’ENMG montre des tracés neurogènes dans les muscles innervés par les racines intéressées.
3 Diagnostic
Il est posé par :
- • une sérologie positive dans le sang et le LCS (après avoir écarté une sérologie croisée avec le tréponème) avec synthèse intrathécale d’anticorps spécifiques;
- • un résultat positif par une technique ELISA (enzyme-linked immunosorbent assay : dépistage) qui doit être confirmé par un résultat positif en Western blot.
4 125Diagnostic différentiel en cas de méningoradiculite non infectieuse
Infiltration méningée au cours d’une pathologie hématologique ou d’une méningite carcinomateuse.
5 Traitement
L’antibiothérapie parentérale par ceftriaxone (2 g par jour pendant 15 à 21 jours consécutifs) permet une régression rapide des manifestations, principalement des douleurs dans un premier temps.
E Méningoradiculites infiltratives
- • Leucémie et lymphomes peuvent être révélés par une atteinte radiculaire avec infiltration méningée.
- • Le diagnostic sera évoqué sur l’atteinte rapidement progressive et étagée : atteinte radiculaire asymétrique des membres inférieurs, des membres supérieurs et des nerfs crâniens.
- • Le diagnostic est confirmé par l’étude du LCS (cytométrie de flux, dosages des cytokines IL6 et IL10) et par le bilan systémique de l’hémopathie.
III Atteintes plexiques (plexopathies)
Les plexopathies sont les lésions des plexus nerveux. Les atteintes du plexus brachial sont les plus fréquentes.
Elles sont diagnostiquées sur une atteinte clinique et neurophysiologique sensitivo-motrice périphérique non systématisable en termes de troncs nerveux ou d’atteinte radiculaire.
A Plexopathie infiltrative
Le syndrome de Pancoast-Tobias correspond à la compression et l’infiltration du plexus cervical inférieur par une tumeur de l’apex pulmonaire. Elle est responsable d’une atteinte prédominant sur les territoires C8-T1, avec douleurs importantes, et d’un signe de Claude Bernard-Horner.
B Plexopathies post-radiques
 Elles surviennent plusieurs années après une irradiation sus-claviculaire ou axillaire dans les cancers ORL, du sein et les lymphomes hodgkiniens ou non. Elles sont d’évolution lentement progressive, indolores, asymétriques le plus souvent, avec un déficit sensitif et moteur à prédominance motrice, avec une aréflexie ostéotendineuse. Des myokymies s’observent habituellement dans les territoires touchés (trémulation musculaire répétée n’induisant pas de mouvement).
Elles surviennent plusieurs années après une irradiation sus-claviculaire ou axillaire dans les cancers ORL, du sein et les lymphomes hodgkiniens ou non. Elles sont d’évolution lentement progressive, indolores, asymétriques le plus souvent, avec un déficit sensitif et moteur à prédominance motrice, avec une aréflexie ostéotendineuse. Des myokymies s’observent habituellement dans les territoires touchés (trémulation musculaire répétée n’induisant pas de mouvement).
Item 96 – Neuropathies périphériques
- I Diagnostiquer les différentes formes de neuropathies périphériques et connaître l’orientation étiologique
- II Distinguer cliniquement une neuropathie périphérique et une sclérose latérale amyotrophique
I Diagnostiquer les différentes formes de neuropathies périphériques et connaître l’orientation étiologique
Le diagnostic d’une neuropathie périphérique nécessite une démarche structurée comportant les étapes suivantes :
- • localisation : consistant à déterminer le caractère tronculaire, radiculaire, polyradiculaire, plexique ou neuronal (atteinte du corps cellulaire);
- • mécanisme physiopathologique : consistant à déterminer le caractère démyélinisant, axonal ou neuronal de la neuropathie;
- • 128classement électroclinique : synthèse des deux précédentes étapes;
- • recherche de la cause : les causes principales des neuropathies sont relativement limitées à partir de la classification électroclinique;
- • approche thérapeutique : elle dépend de la cause retenue.
L’arbre décisionnel de la figure 8.9 fait la synthèse de cette démarche.
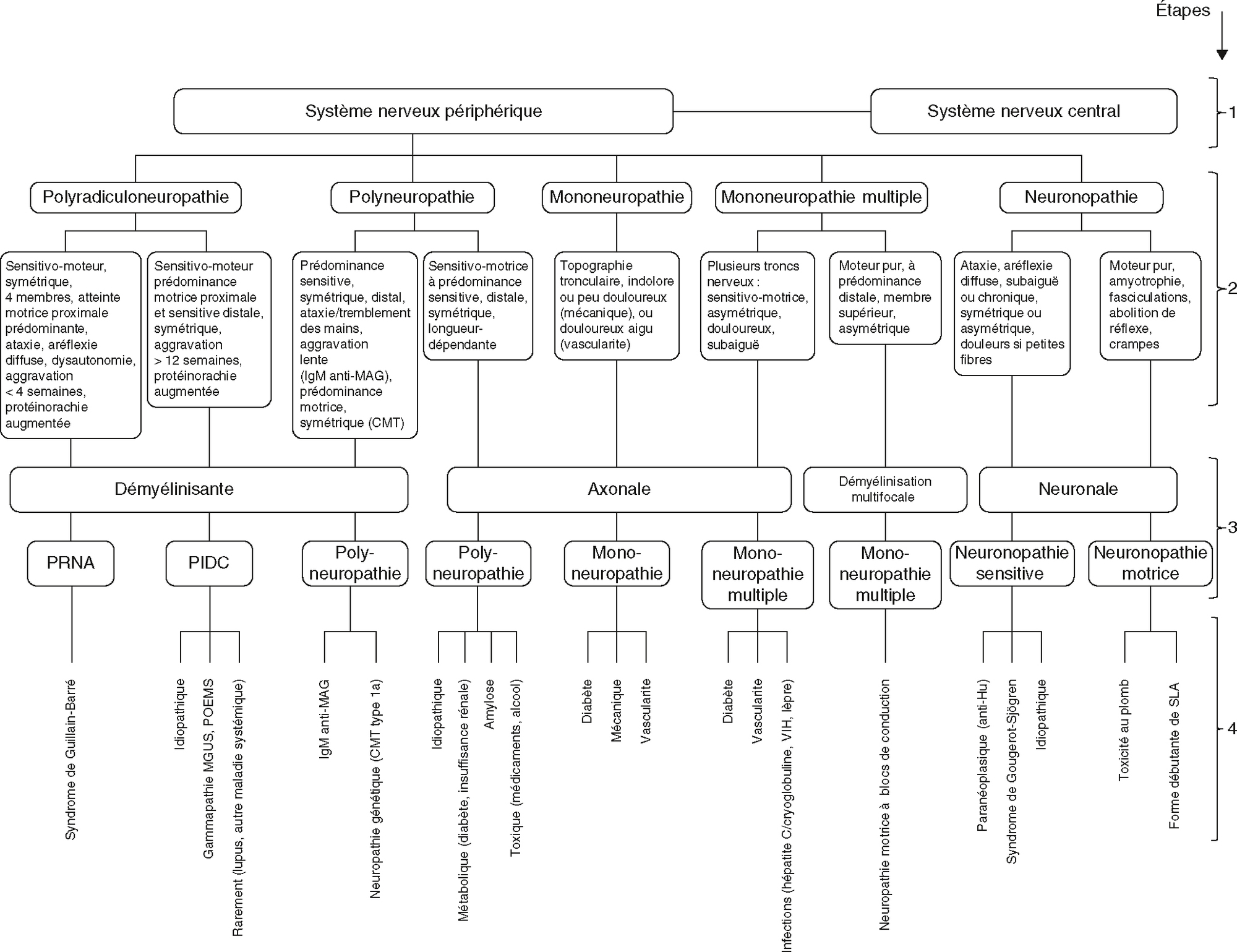
Arbre décisionnel pour le diagnostic des neuropathies périphériques. La lecture des étapes de l’arbre décisionnel s’effectue du haut vers le bas. 1. La symptomatologie et l’examen clinique différencient une atteinte du système nerveux périphérique de celle du système nerveux central. 2. À l’aide des principales caractéristiques (encadrés avec description) la neuropathie sera classée selon un des cinq grands tableaux cliniques. 3, 4. L’examen neurophysiologique permet : (3) une classification électro-clinique et (4) de réduire le nombre d’hypothèses sur la cause de la neuropathie. À noter que la neuropathie motrice à blocs de conduction persistants est une mononeuropathie multiple dont le mécanisme dysimmunitaire est repon-sable de blocs de conduction en rapport avec des zones de démyélinisation focales (segmentaires). CMT : maladie de Charcot-Marie-Thooth; IgM : immunoglulines M; MAG : myelin-associatedglycoprotein ; MGUS : monoclonal gammopathy of undetermined significance ; POEMS : Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal plasma cell disorder, Skin Changes ; PRNA : polyradiculonévrite aiguë; PIDC : polyneuropathie démyélinisante inflammatoire chronique; SLA : sclérose latérale amyotrophique; VIH : virus de l’immunodéficience humaine. Remerciements à Alain Créange, CHU Henri-Mondor.
Organisé de manière hiérarchique, cette décision permet aux décisions d'identifier diverses formes de neuropathie périphérique, à travers la distribution anatomique de l'attaque et la nature de la lésion. La distinction entre le polyradiculonisme, multiple, monogène, polymyosinflammatoire et la névrose est réalisée ci-dessus. Toutes les entités sont alors basées sur des mécanismes sous-jacents tels que la démyélinisation, les axones, les points multifocaux et les neurones. La présence d'une table multifocale asymétrique, ou avec un avantage distal ou proximal, provoque des formes mixtes ou compliquées. Ce voyage visuel encouragera une lecture progressive et logique pour guider les recherches supplémentaires et établir un diagnostic plus précis.
129En matière d’introduction, rappelons la classification et la définition des neuropathies périphériques :
- • mononeuropathie : atteinte d’un tronc nerveux;
- • mononeuropathie multiple : atteinte de plusieurs troncs nerveux (multinévrite);
- • polyneuropathie : atteinte de toutes les fibres nerveuses, en fonction de leur longueur (atteinte longueur-dépendante);
- • polyradiculoneuropathie : atteinte de l’ensemble des racines nerveuses sensitives et motrices et des troncs nerveux (exemple du syndrome de Guillain-Barré);
- • neuronopathie : atteinte du corps cellulaire du neurone (moteur ou sensitif; terme le plus souvent employé pour les neuronopathies sensitives).
A Diagnostic positif
1 Clinique
Le diagnostic repose sur l’association plus ou moins complète de trois ordres de signes.
a Signes moteurs
Les signes moteurs associent :
- • une paralysie (déficit complet), ou une parésie (déficit incomplet), flasque; elle est cotée de 0 à 5 (testing musculaire)1;
- • une amyotrophie (retardée de 3 semaines par rapport à la lésion nerveuse en cas d’atteinte aiguë);
- • des fasciculations (spontanées ou provoquées par la percussion ou l’exposition au froid); traduisant l’activité spontanée d’une unité motrice, elles sont en faveur d’une lésion proche de la corne antérieure (motoneurone, racine);
- • des crampes au repos;
- • une diminution ou une abolition des réflexes tendineux.
b Signes sensitifs
Les signes sensitifs sont souvent les premiers à apparaître.
Signes subjectifs
- • Ces sensations anormales sont :
-
- – des paresthésies (picotements, fourmillements, engourdissements spontanés);
- – des dysesthésies (déclenchées par le tact, le frottement : allodynie, douleur provoquée par un stimulus habituellement non douloureux);
- – des douleurs (brûlures, décharges électriques, striction).
- • La topographie des manifestations douloureuses ou non peut être longueur-dépendante (polyneuropathie) ou tronculaire :
-
- – les manifestations sont symétriques distales (aux pieds puis ascendantes) dans le cadre d’une polyneuropathie;
- – les manifestations tronculaires (d’un nerf mixte) sont limitées au territoire du tronc nerveux, le plus souvent sur une zone limitée de la main ou du pied, sans dépasser le poignet ou la cheville.
- • 130À différencier des manifestations radiculaires :
-
- – la douleur radiculaire a un trajet traçant le long d’un membre, augmentée par les manœuvres élevant la pression du LCS (toux, éternuement) et celles étirant la racine (signe de Lasègue, mouvements du cou) en cas d’atteinte mécanique; les signes objectifs sont absents ou discrets.
Atteinte sensitive objective
- • Les troubles concernent les sensibilités superficielle (au tact, ou sensibilité épicritique; à la température et à la piqûre, ou sensibilité thermoalgique) et proprioceptive (altération du sens de position des segments de membre, avec ataxie et signe de Romberg).
- • L’ataxie est évocatrice d’une neuropathie démyélinisante.
c Signes neurovégétatifs
Il s’agit :
- • des signes vasomoteurs (œdème, cyanose);
- • des troubles trophiques (peau sèche, squameuse et atrophique) et des phanères (chute de poils, ongles cassants);
- • d’une hypotension orthostatique;
- • d’une impuissance;
- • des troubles digestifs (diarrhée, constipation, gastroparésie), d’une incontinence urinaire;
- • d’un trouble de la motilité pupillaire.
d Autres signes
Ils comportent la recherche de gros nerfs et d’un signe de Tinel (douleur projetée sur le trajet du nerf lors de la percussion de celui-ci).
On peut, à l’issue des données cliniques, distinguer différents types d’atteintes (fig. 8.2) :
- • atteinte des grosses fibres (myélinisées) : troubles des sensibilités proprioceptive et tactile, signes moteurs;
- • atteinte des petites fibres (amyéliniques) : troubles de la sensibilité thermoalgique et manifestations neurovégétatives – cette dernière association n’est pas systématique.
2 Examens complémentaires
a 131Électrophysiologie neuromusculaire (ou électroneuromyographie)
L’électroneuromyographie (ENMG) confirme la nature neurogène du trouble. Elle détermine le mécanisme démyélinisant, axonal ou neuronal.
L’examen comporte deux temps distincts.
Électromyogramme (détection)
L’électromyogramme (analyse à l’aiguille des tracés de repos et en cours de contraction des différents muscles étudiés) montre le caractère neurogène des anomalies : potentiels de fibrillation au repos, appauvrissement du nombre d’unités motrices, accélération de la fréquence des potentiels individualisés (sommation temporelle) et, au maximum, potentiel à fréquence élevée (tracé simple) lors de la contraction (fig. 8.10).
Il donne une indication topographique (tronculaire, radiculaire, diffuse) en fonction des muscles touchés et confirme l’absence de signes myogènes (richesse exagérée des tracés pour un faible effort de contraction, potentiels de faible amplitude très polyphasiques).
Mesure de la conduction nerveuse (examen de stimulodétection)
Elle reflète le mécanisme physiopathologique :
- • neuropathies démyélinisantes :
-
-
– ralentissement des vitesses de conduction motrices (fig. 8.11),
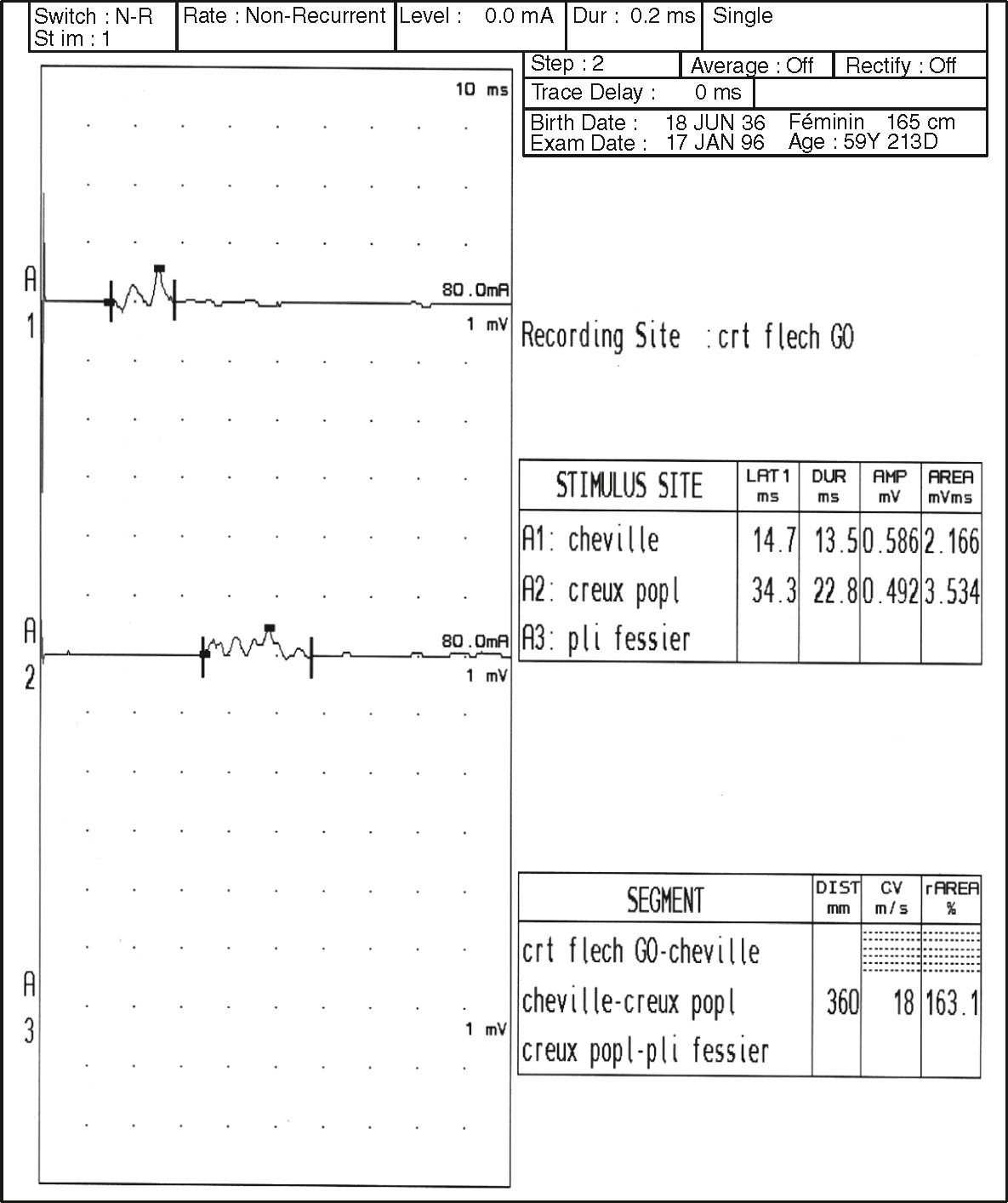
Fig. 8.11 Vitesse de conduction ralentie (vitesse de conduction à 18 m/s) et allongement de la latence distale (Lat A1 = 14,7 ms) dans le nerf fibulaire.Ce tracé de conduction nerveuse motrice montre un enregistrement réalisé au niveau du muscle court fléchisseur des orteils gauches (crt flech GO) suite à une stimulation en trois points distincts du nerf fibulaire : la cheville (A1), le creux poplité (A2) et le pli fessier (A3). Les données révèlent une latence distale augmentée à 14,7 ms au niveau de la cheville, ce qui est significativement prolongé, traduisant un retard de conduction. De plus, la vitesse de conduction mesurée entre le creux poplité et le pli fessier est fortement ralentie à 18 m/s, bien en dessous des valeurs normales attendues. L’ensemble du profil électrophysiologique indique une altération de la conduction motrice du nerf fibulaire, compatible avec une neuropathie périphérique d’allure démyélinisante ou compressive.
- – allongement des latences des ondes F,
- – allongement des latences distales motrices,
- – dispersion des potentiels d’action,
- – blocs de conduction motrice (rapport diminué entre l’amplitude obtenue par stimulation proximale et celle obtenue par stimulation distale), sans augmentation de la durée du potentiel : on peut les observer au cours des polyradiculonévrites, des neuropathies à blocs de conduction persistants et des neuropathies canalaires (fig. 8.12);
-
- • 132neuropathies axonales :
-
- – vitesses normales (ou modérément ralenties),
- – baisse d’amplitude du potentiel d’action de la réponse motrice,
- – baisse d’amplitude du potentiel d’action de la réponse sensitive;
- • neuronopathies sensitives :
-
- – baisse d’amplitude ou abolition diffuse (aux quatre membres) du potentiel d’action sensitif,
- – normalité des paramètres moteurs;
- • neuronopathies motrices :
-
- – baisse d’amplitude du potentiel d’action moteur, de topographie variable suivant la cause,
- – normalité des paramètres sensitifs.
b 133Liquide cérébrospinal
- • Hyperprotéinorachie isolée dans les polyradiculonévrites, le diabète.
- • Réaction cellulaire dans certaines affections paranéoplasiques.
- • Véritable méningite en cas de pathologies néoplasiques et hématologiques.
c Biopsie du nerf sensitif
- • La biopsie de nerf sensitif permet dans certains cas difficiles de confirmer le mécanisme de la neuropathie (démyélinisation segmentaire, dégénérescence axonale) et oriente le diagnostic étiologique.
- • Elle conduit à amputer un nerf sensitif. Elle n’est donc jamais pratiquée en première intention.
- • Elle est réalisée le plus souvent sur un nerf sensitif de la jambe (branche sensitive du nerf fibulaire ou nerf sural).
- • Elle permet de mettre en évidence des arguments diagnostiques :
-
- – occlusion artérielle en cas de vascularite et perte axonale fasciculaire;
- – dépôts d’amylose en cas de neuropathie amyloïde;
- – lésions inflammatoires et démyélinisation/remyélinisation d’une polyneuropathie inflammatoire démyélinisante chronique.
B Diagnostic différentiel
II est développé au chapitre 7 (item 92), ainsi que plus loin dans la partie II. Distinguer clini-quement une neuropathie périphérique et une sclérose latérale amyotrophique.
1 134Paralysies centrales
Il s’agit de paralysies spastiques, non amyotrophiantes.
2 Atteintes myogènes
Ce sont des atteintes motrices pures le plus souvent proximales, fréquemment bilatérales, avec abolition de la réponse idiomusculaire (éléments du diagnostic : données électromyogra-phiques, élévation du taux des enzymes musculaires). Absence de systématisation tronculaire ou radiculaire de l’atteinte.
3 Sclérose latérale amyotrophique
Voir plus loin II. Distinguer cliniquement une neuropathie périphérique et une sclérose latérale amyotrophique.
C Orientation du diagnostic étiologique
1 Classification électroclinique
- • Mononeuropathie : atteinte d’un tronc nerveux (nerf médian, nerf ulnaire, nerf fibulaire) responsable d’une atteinte sensitivo-motrice.
- • Mononeuropathie multiple : atteinte de plusieurs troncs nerveux.
- • Polyneuropathie :
-
- – atteinte de toutes les fibres nerveuses en fonction de leur longueur (atteinte longueur-dépendante, c’est-à-dire commençant par une atteinte distale des membres inférieurs, en raison de lésions des fibres les plus longues);
- – à prédominance sensitive;
- – d’évolution souvent chronique;
- – ataxiante lorsqu’elle est démyélinisante.
- • Polyradiculoneuropathie :
-
- – atteinte de l’ensemble des racines nerveuses sensitives et motrices et des troncs nerveux : c’est l’exemple du syndrome de Guillain-Barré, voir plus loin Item 97 – Polyradiculonévrite aiguë inflammatoire (syndrome de Guillain-Barré);
- – atteinte relativement symétrique;
- – sensitive et motrice;
- – avec une composante proximale;
- – une aréflexie diffuse habituellement;
- – une augmentation de la protéinorachie;
- – pouvant être aiguë ou chronique.
- • Neuronopathie :
-
- – atteinte du corps cellulaire du neurone (moteur ou sensitif);
- – les neuronopathies sensitives peuvent avoir plusieurs présentations; la plus caractéristique est l’association de :
-
- – troubles sensitifs asymétriques non longueur-dépendants touchant les membres (de façon caractéristique, simultanément les membres supérieurs et les membres inférieurs), le tronc ou la face,
- – aréflexie diffuse,
- – ataxie,
- – 135hyperprotéinorachie avec réaction cellulaire en cas d’origine paranéoplasique,
- – elles peuvent prendre le masque d’une polyneuropathie sur le plan clinique, le diagnostic étant attesté par l’abolition diffuse et isolée des potentiels sensitifs sur l’examen neurophysiologique, et confortée par l’hyperprotéinorachie;
- – les neuronopathies motrices sont souvent asymétriques; elles sont un mode d’entrée fréquent dans la sclérose latérale amyotrophique et ont alors une distribution qui n’est ni tronculaire ni radiculaire mais plutôt métamérique (voir plus loin II. Distinguer cliniquement une neuropathie périphérique et une sclérose latérale amyotrophique).
2 Orientation étiologique
Elle nécessite au préalable :
- • une démarche clinique :
-
- – la recherche de formes familiales,
- – la recherche des prises médicamenteuses neurotoxiques;
- • un examen neurophysiologique (ENMG);
- • parfois un examen du LCS;
- • une approche biologique minimale comprenant : numération formule sanguine (NFS), vitesse de sédimentation (VS)-C reactiveprotein (CRP), glycémie à jeun et 2 heures après la prise de 75 g de glucose per os, ionogramme, bilan hépatique, hémostase, immunofixation des immunoglobulines du sérum à la recherche d’une gammapathie monoclonale.
D Polyneuropathies axonales longueur-dépendantes
Elles résultent d’une atteinte diffuse et symétrique intéressant les extrémités distales des fibres nerveuses.
Une polyneuropathie débute donc aux pieds. Lorsqu’elle atteint les genoux, les manifestations touchent alors les mains. Lorsqu’elle atteint les coudes, les fibres de l’abdomen (plastron) et du scalp (atteinte en calotte) sont touchées à leur tour (fig. 8.13).
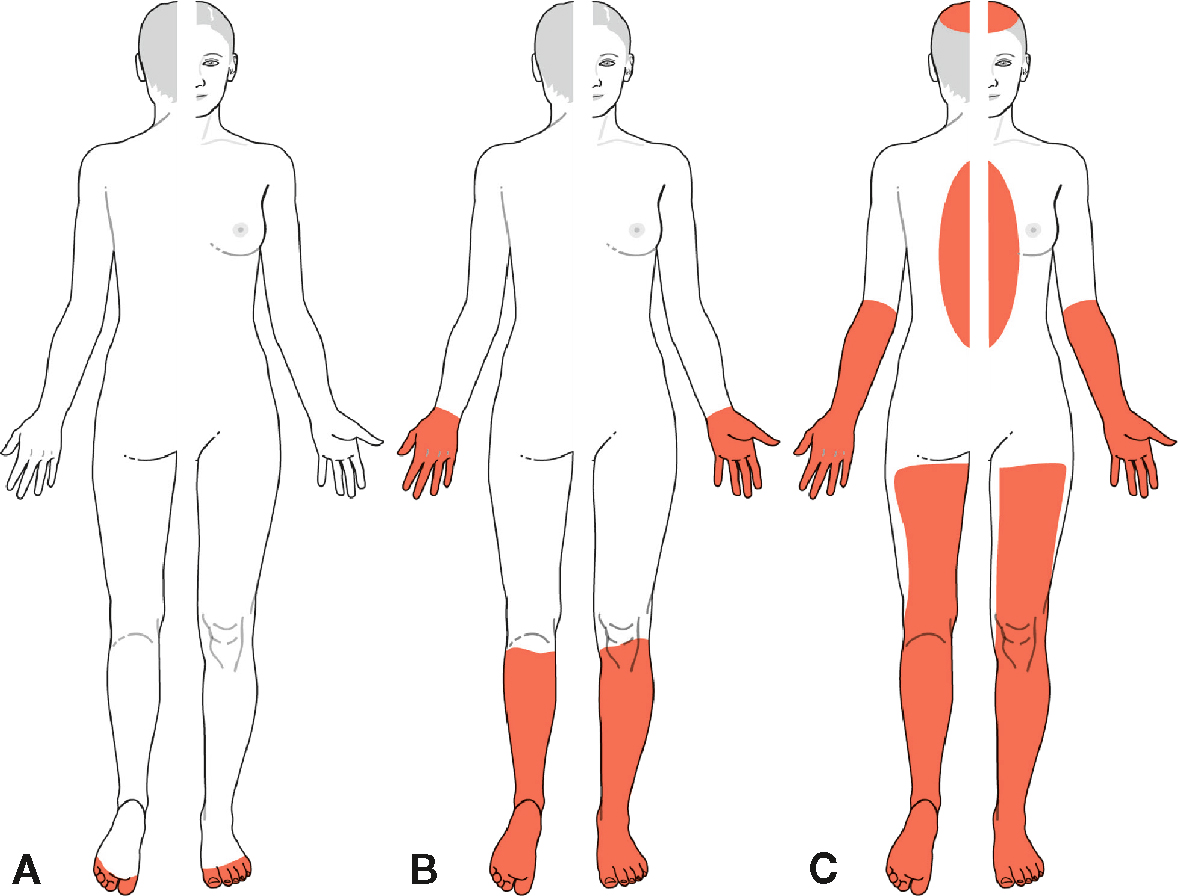
Topographie des manifestations neurologiques dans les polyneuropathies. A. Forme initiale de polyneuropathie. Atteinte distale symétrique des fibres les plus longues aux pieds. B. Atteinte longueur-dépendante, distale et symétrique. Typiquement, les troubles commencent à atteindre les mains lorsque, aux membres inférieurs, les troubles s’étendent aux genoux. C. Extension de la polyneuropathie, les fibres plus courtes sont atteintes, responsables d’une atteinte en plastron (abdomen et face antérieure du thorax) et en calotte (scalp).
Illustration représentant la distribution typique des troubles sensitifs dans les polyneuropathies, selon différentes présentations cliniques. La figure A montre une atteinte distale symétrique limitée aux extrémités, avec un début aux pieds, fréquent dans les neuropathies périphériques débutantes. La figure B illustre une atteinte en « gants et chaussettes », classique des polyneuropathies axonales ou métaboliques évoluées, où les déficits sensoriels touchent symétriquement les mains et les pieds. La figure C met en évidence une atteinte disséminée plus étendue, avec participation du tronc et du visage, retrouvée dans certaines polyneuropathies inflammatoires ou auto-immunes. La coloration rouge délimite les zones corporelles affectées par les manifestations neurologiques.
1 Clinique
a Troubles sensitifs
Souvent initiaux, les troubles subjectifs (paresthésies permanentes, dysesthésies, brûlures, allo-dynie mécanique), touchant les extrémités des membres inférieurs sont permanents, diurnes et volontiers nocturnes. L’atteinte des grosses fibres myélinisées se traduit par des troubles de la sensibilité profonde responsables d’une ataxie.
b Troubles moteurs
Au début, difficultés à la marche, fatigabilité anormale; puis s’installe un steppage (déficit symétrique des releveurs du pied). Il n’y a pas d’atteinte des muscles respiratoires ni des nerfs crâniens. Il existe parfois des crampes (mollets, plante des pieds).
c Troubles végétatifs
Ils sont liés à une atteinte des petites fibres amyéliniques : hypotension artérielle orthostatique, troubles vésicosphinctériens, sexuels (impuissance), troubles digestifs (diarrhée, constipation), dépilation et troubles vaso-moteurs distaux. Leur présence évoque certaines causes telles que le diabète et l’amylose.
d 136Examen clinique
Il confirme l’atteinte symétrique, à prédominance distale, des membres inférieurs : abolition des réflexes calcanéens (achilléens), déficit moteur affectant les releveurs du pied, amyotrophie, déficit sensitif (au tact ou à la piqûre), atteinte végétative (peau et phanères, hypotension).
e Démarche étiologique
Elle dépend de nombreux facteurs :
- • âge : enfant (rare et héréditaire), adulte (très nombreuses causes);
- • origine : amylose familiale (dont un foyer important est situé au Portugal);
- • circonstances : maladie générale (diabète, insuffisance rénale), médicaments;
- • mode d’installation : toujours déterminant (aigu, subaigu, chronique).
2 Causes des polyneuropathies axonales
Il faut rechercher une cause générale susceptible d’altérer la physiologie de l’axone. On retiendra donc essentiellement les causes métaboliques et toxiques (dont les causes médicamenteuses), sans oublier l’amylose.
a Neuropathie diabétique
Le diabète est une des causes les plus fréquentes de neuropathie périphérique. L’incidence est difficile à estimer, pouvant atteindre 60 %. Sa répartition est égale dans les deux sexes.
137 Plusieurs mécanismes sont en cause : facteurs vasculaires (augmentation de la perméabilité des capillaires endoneuraux), facteurs métaboliques (accumulation de sorbitol), facteurs inflammatoires (infiltrats de lymphocytes et de macrophages dans les fascicules des fibres amyéliniques).
Il existe très probablement un lien entre l’ancienneté de l’hyperglycémie et les neuropathies, qui apparaissent le plus souvent 5 à 10 ans après le début du diabète. De plus, les traitements qui maintiennent une glycémie relativement normale peuvent stabiliser, voire améliorer certaines anomalies neurologiques (dans le diabète de type 1).
Polyneuropathie sensitivo-motrice distale
- • Forme la plus souvent observée des neuropathies diabétiques, associée ou non à une atteinte dysautonomique.
- • Survenant le plus souvent chez des patients dont le diabète évolue depuis plus de 5 ans, le début en est généralement lentement progressif.
- • Il s’agit au début de paresthésies (engourdissements, picotements et brûlures) des pieds. L’examen révèle une aréflexie calcanéenne et une hypoesthésie affectant la sensibilité ther-moalgique « en chaussettes » et la sensibilité vibratoire. Les douleurs sont fréquentes (pieds et jambes), à type de constriction, de broiement avec allodynie mécanique au frottement à l’examen clinique.
- • Les manifestations dysautonomiques dans les formes plus évoluées comportent des troubles cardiovasculaires (hypotension orthostatique, cardiopathie autonome diabétique), des troubles de la sphère digestive (constipation, douleurs abdominales, nausées, dyspha-gie, diarrhée et incontinence fécale), des troubles génito-urinaires (impuissance, atonie vésicale), des troubles de la motilité pupillaire et une anhydrose fréquente.
Autres neuropathies diabétiques
Il ne s’agit plus alors de polyneuropathies.
Neuropathies motrices proximales (ou amyotrophie diabétique)
Elles s’installent de façon subaiguë sur quelques jours à semaines : faiblesse musculaire avec amyotrophie des racines des membres inférieurs asymétrique, qui concerne électivement le psoas, les quadriceps, les adducteurs et les muscles postérieurs de cuisse. Des douleurs importantes sont présentes dans les territoires concernés. Les réflexes tendineux sont diminués ou abolis aux membres inférieurs et il n’existe pas ou peu de signes sensitifs déficitaires.
Neuropathies focales et multifocales
Elles comportent :
- • les atteintes des nerfs crâniens (surtout oculomoteurs, nerf facial);
- • les atteintes des membres (tous les troncs nerveux, mais surtout le nerf ulnaire, le nerf médian et le nerf fibulaire commun);
- • l’atteinte du nerf fémoral reste la plus évocatrice : début aigu avec douleurs à type d’écrasement et à recrudescence nocturne, déficit quadricipital amyotrophiant, abolition du réflexe rotulien, déficit sensitif dans le territoire du nerf fémoral (correspondant à une forme d’amyotrophie diabétique tronculaire) (voir plus haut).
Neuropathies du tronc
Douleurs de la poitrine et/ou de l’abdomen de topographie tronculaire (nerf intercostal).
Traitement
A Il repose sur l’équilibre du diabète, le contrôle de la douleur, le traitement de la dysautono-mie (hypotension orthostatique, troubles sphinctériens et du transit intestinal) ainsi que le contrôle du syndrome métabolique, souvent associé en cas de diabète de type 2.
b 138Causes médicamenteuses et toxiques
- • Les causes médicamenteuses sont les plus fréquentes :
-
- – il s’agit le plus souvent de polyneuropathies sensitives axonales, parfois douloureuses;
- – les substances le plus souvent incriminées sont :
-
- – les cytostatiques (notamment la vincristine et les platines : neuronopathies),
- – la thalidomide (nécessitant une surveillance systématique),
- – l’isoniazide,
- – l’amiodarone,
- – la nitrofurantoïne, les antirétroviraux,
- – le disulfirame, la chloroquine, le métronidazole;
- – A l’ENMG peut être utilisé pour dépister les manifestations infracliniques, notamment pour permettre la poursuite de certaines prescriptions (thalidomide).
- • Les causes toxiques sont principalement d’origine industrielle, comme le benzène. Elles sont plus rares et peuvent se révéler de manière aiguë.
c Polyneuropathie toxique alcoolique
Voir chapitre 2 – item 76.
- • Deuxième cause de polyneuropathie dans les pays industrialisés après le diabète, elle affecterait plus de 10 % des alcooliques chroniques. Elle est habituellement secondaire à la toxicité directe de l’alcool. L’association à une carence en thiamine (vitamine B1), avec ou sans carence en folates associée, est possible. Elle touche les fibres motrices, sensitives et végétatives. La symptomatologie sensitive est prédominante.
- • S’installant de façon insidieuse et lentement progressive, la polyneuropathie liée à l’alcoolisme chronique se traduit au début par des paresthésies à type de fourmillements des pieds et « en chaussettes », des crampes nocturnes des mollets, une faiblesse motrice s’expri-mant par une fatigabilité anormale à la marche. Après un certain temps d’évolution, le patient se plaint de douleurs en étau et surtout de brûlures avec paroxysmes, voire d’une hyperpathie douloureuse, surtout nocturne.
- • Des troubles cutanés et trophiques (dépilation, anhydrose, ongles cassants) sont fréquemment associés.
- • L’examen clinique met en évidence une hypoesthésie symétrique, « en chaussettes », des différents types de sensibilité, moins marquée pour la sensibilité proprioceptive.
- • Il révèle une aréflexie calcanéenne, une amyotrophie et un déficit moteur prédominant sur les muscles de la loge antérolatérale de jambe.
- • ENMG : les anomalies sont très précoces, axonales.
Traitement
- • Il associe l’administration parentérale de vitamines, surtout B1, en cas de carence et de sevrage, et un régime riche en protéines.
- • En cas de douleurs importantes, l’utilisation de tricycliques et/ou d’antiépileptiques.
- • Cette thérapeutique, associée à l’arrêt de l’intoxication, permet une amélioration clinique et électrophysiologique, mais celle-ci peut s’étendre sur plusieurs mois.
Autres formes cliniques
- • Une polyneuropathie aiguë, responsable d’une paraparésie flasque amyotrophiante, de troubles sensitifs intéressant toutes les modalités et d’une aréflexie rapidement ascendante en 24 heures, peut survenir chez l’alcoolique dénutri, volontiers à la suite d’un état infectieux en raison d’une carence associée en vitamine B1.
- • 139Une forme ulcéromutilante comportant un déficit thermoalgique sévère, des maux perforants plantaires avec ostéolyse, arthropathies, a été individualisée.
- • Des formes végétatives, le plus souvent associées à la polyneuropathie axonale, comportant des troubles de sudation, une hypotension orthostatique, une impuissance, des troubles trophiques et digestifs, sont de pronostic plus réservé, notamment lorsque sont associés des troubles du rythme cardiaque.
d Amylose héréditaire et primitive (gammapathie)
La polyneuropathie amyloïde est secondaire aux dépôts de substance amyloïde (préalbumine mutée au cours des amyloses héréditaires ou gammapathie monoclonale au cours des amy-loses primitives) dans le nerf.
Initialement marquées par un déficit de la sensibilité thermoalgique pouvant s’associer à des douleurs, les manifestations s’étendent ensuite aux membres supérieurs, au tronc avec des manifestations dysautonomiques.
Diagnostic :
- • il est suggéré par le contexte familial (pas toujours présent);
- • il est réalisé par la mise en évidence de la substance amyloïde et par le test génétique à la recherche d’une mutation sur le gène de la transthyrétine (préalbumine); la plus fréquente est la mutation VAL/MET30 :
-
- – dans la biopsie des glandes salivaires accessoires, la graisse abdominale ou la biopsie anale,
- – dans la biopsie du nerf sensitif.
e Vascularite
Vingt à 30 % des vascularites prennent la forme clinique d’une polyneuropathie, même si le mécanisme est vasculaire. Une mononeuropathie multiple est plus évocatrice. Lorsque la neuropathie est évoluée, l’asymétrie disparaît. Il importe de rechercher dans l’histoire clinique un début asymétrique aux membres inférieurs, avec atteinte du nerf fibulaire le plus souvent.
Il s’agit d’une polyneuropathie plutôt subaiguë avec composante motrice et douloureuse.
f Causes infectieuses
Le SIDA peut entraîner une polyneuropathie sensitive à la phase tardive de l’affection.
g Autres causes
D’autres causes plus rares de polyneuropathies axonales sont possibles, telles que les hypothyroïdies.
E 140Principales polyneuropathies démyélinisantes
1 Neuropathie à IgM monoclonale à activité anti-MAG
- • Neuropathie de la personne de plus de 50 ans.
- • Ataxiante, associée à un tremblement des mains.
- • Peu de déficit moteur au début.
- • Lentement progressive.
- • L’ENMG montre une polyneuropathie démyélinisante à prédominance distale.
- • Biologie : pic d’IgM monoclonale dont le taux est faible, inférieur à 10 g/L (plus rarement supérieur à 10 g/L dans le cadre d’une maladie de Waldenström).
- • Présence d’anticorps anti-MAG (myelin-associated glycoprotein, constituant de la myéline intervenant dans la compaction de cette dernière).
2 Neuropathies héréditaires
Les neuropathies héréditaires sont dominées par les polyneuropathies sensitivo-motrices de Charcot-Marie-Tooth : atrophie des mollets, pieds creux (fig. 8.14), atteinte sensitivo-motrice à prédominance motrice. Peu ou pas de symptomatologie sensitive (paresthésies).
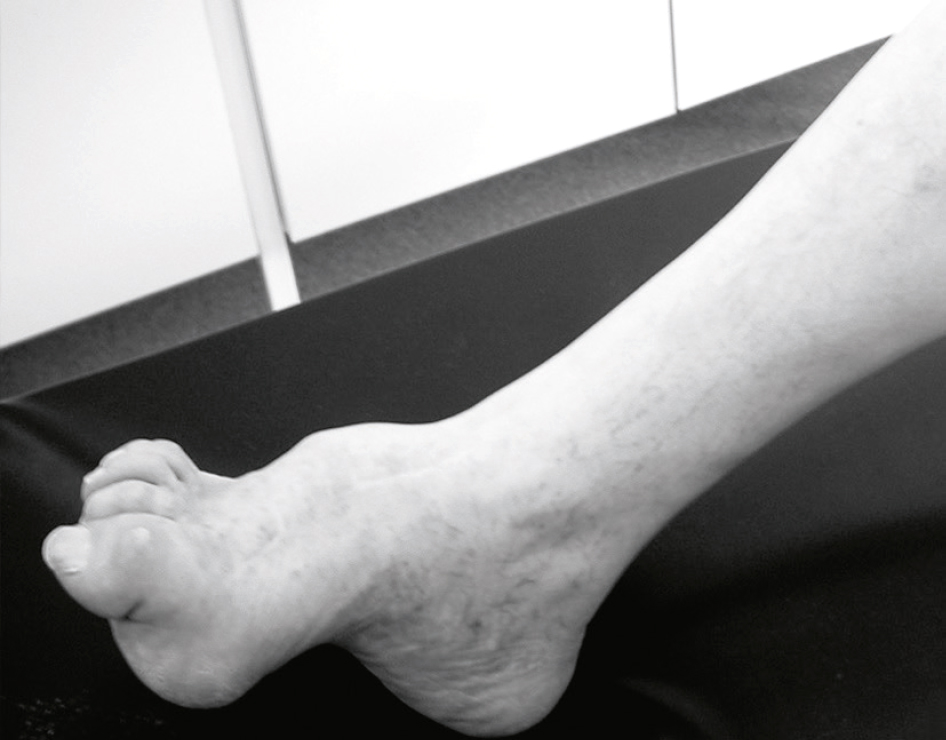
Pied creux (neuropathie de Charcot-Marie-Tooth).Cette photographie montre un pied atteint de déformation en pied creux, observé chez un patient présentant une neuropathie héréditaire de type Charcot-Marie-Tooth. L’arche plantaire est exagérément accentuée, traduisant une rétraction des muscles plantaires et un déséquilibre entre les muscles fléchisseurs et extenseurs du pied. On observe également une déviation en griffe des orteils, notamment au niveau du gros orteil, ainsi qu’une inversion du pied, souvent associée à une faiblesse des muscles péroniers latéraux. L’ensemble de la posture du pied révèle une instabilité structurelle et une contracture chronique. La peau apparaît fine, avec une absence visible de masse musculaire au niveau du mollet, suggérant une amyotrophie distale caractéristique de cette pathologie. Cette configuration biomécanique complexe entraîne des troubles de la marche et un risque accru d’entorses ou de douleurs chroniques.
Les formes démyélinisantes autosomiques dominantes sont les mieux caractérisées. Le diagnostic est porté par le test génétique : duplication du gène PMP22 sur le chromosome 17 dans les formes dominantes, anomalie du gène de la connexine 32 dans les formes liées à l’X.
F 141Neuronopathies sensitives
1 Présentations cliniques des neuronopathies sensitives (vidéo 8.3)
-
• A Les neuronopathies sensitives correspondent à la dégénérescence du corps cellulaire des neurones sensitifs dans la racine postérieure des nerfs spinaux (fig. 8.15).
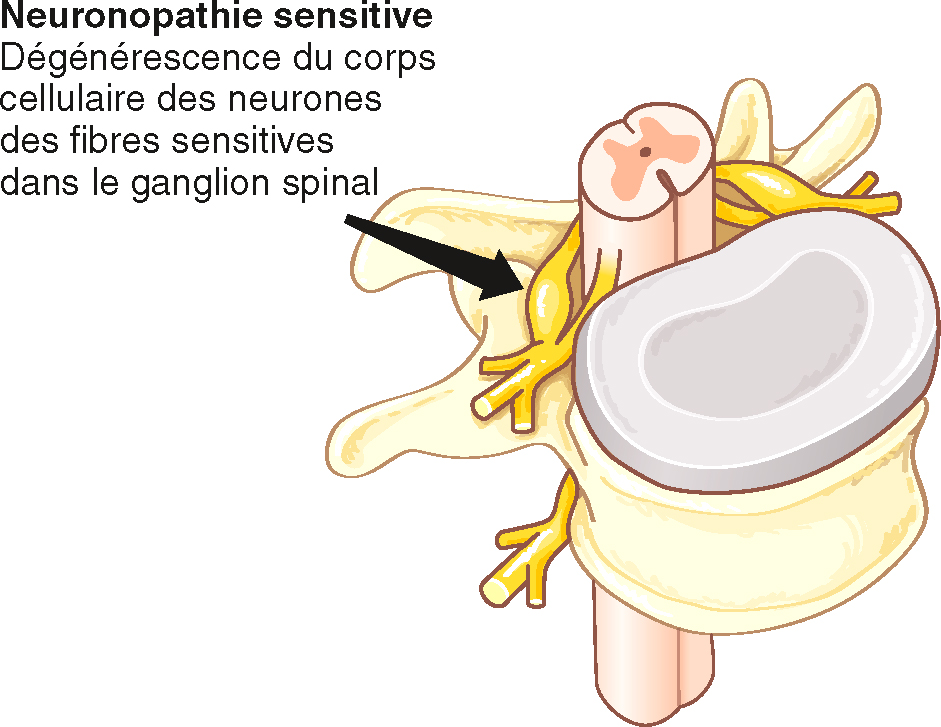
Fig. 8.15 La lésion au cours des neuronopathies sensitives est située dans le ganglion spinal, correspondant à la localisation des corps cellulaires des neurones sensitifs périphériques.
Illustration de Carole Fumat.Illustration anatomique d’un segment rachidien montrant la localisation précise de la lésion dans les neuronopathies sensitives. L’image met en évidence la dégénérescence des corps cellulaires des neurones sensitifs situés au niveau du ganglion spinal, représenté ici comme un renflement sur la racine postérieure du nerf spinal. Une flèche noire pointe directement vers cette structure, soulignant son implication dans la pathologie. La moelle épinière, en coupe transversale, est entourée des arcs vertébraux et du disque intervertébral, avec les racines nerveuses sortant latéralement. L’image met ainsi en valeur que, contrairement à une atteinte radiculaire classique, la lésion neuronopathique se situe en amont, au niveau du ganglion, entraînant une atteinte bilatérale, non systématisée et non dermatomérique de la sensibilité, ce qui est typique de cette entité clinique. La structure osseuse et les nerfs émergents sont représentés avec un rendu tridimensionnel simplifié, facilitant la compréhension du siège de la dégénérescence.
- • Elles sont caractérisées par des manifestations exclusivement sensitives.
-
• L’atteinte est typiquement asymétrique et non systématisée en termes de tronc ou de racines, et concerne les quatre membres, voire la face (25 % des cas), de manière synchrone ou asynchrone (fig. 8.16).
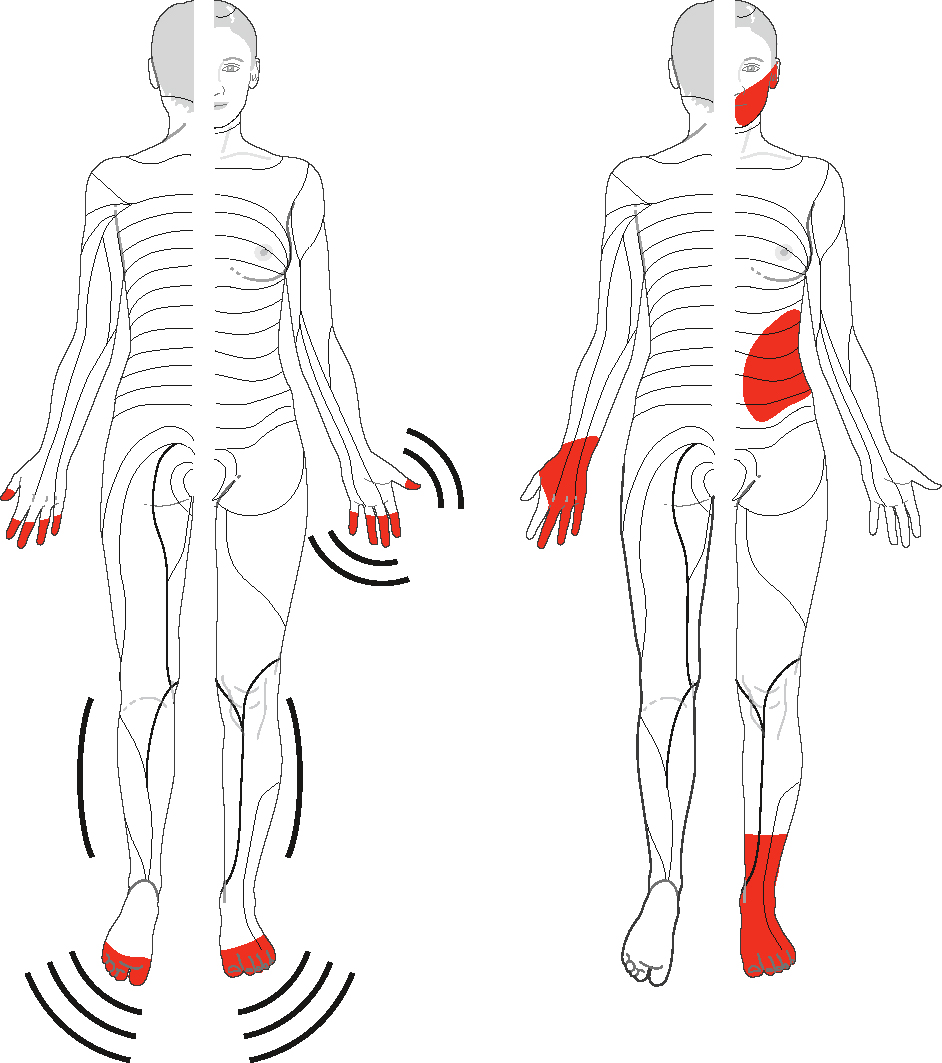
Fig. 8.16 La présentation clinique des neuronopathies sensitives est celle de troubles sensitifs aux quatre membres accompagnés d’ataxie ou de manifestations sensitives multifocales non systématisées en termes de tronc ou de racines.
En rouge : troubles sensitifs; en noir : traits schématisant l’ataxie.
Illustration de Carole Fumat.Schéma anatomique divisé en deux moitiés représentant la répartition corporelle des troubles sensitifs dans le cadre d’une neuronopathie sensitive. Sur la partie gauche, les zones colorées en rouge aux extrémités des mains et des pieds illustrent une atteinte bilatérale et symétrique, accompagnée de traits ondulés noirs suggérant des paresthésies ou des sensations anormales. Sur la partie droite, les zones rouges sont asymétriques et intéressent le visage, une partie du tronc, un bras et une jambe du côté opposé, traduisant une atteinte radiculaire dissociée. Cette juxtaposition met en contraste une distribution distale et diffuse touchant les quatre membres, caractéristique des neuronopathies sensitives, avec une atteinte topographique évoquant un autre type de lésion neurologique. La posture neutre des deux figures et le choix visuel clair permettent une lecture immédiate des territoires atteints, soulignant la particularité des troubles sensoriels associés à ces pathologies.
- • L’ataxie est fréquente.
- • L’aréflexie est diffuse et quasi constante.
- • Les neuronopathies sensitives prédominent, soit sur les grosses fibres (ataxie), soit sur les petites fibres (troubles de la sensibilité thermoalgique).
- • Suivant les causes, leur évolution est chronique ou subaiguë.
- • Elles ont le plus souvent un mécanisme dysimmunitaire.
2 142Diagnostic
- • Il est porté sur l’association d’une atteinte sensitive pure clinique confirmée par une atteinte sensitive pure à l’ENMG : abolition diffuse des potentiels sensitifs sans anomalie des potentiels moteurs.
- • L’analyse du LCS peut montrer :
-
- – une augmentation de la protéinorachie : < 1 g/L non constante;
- – une augmentation du nombre de cellules, qui évoque une origine paranéoplasique.
- • Immunologie : la présence d’anticorps anti-Hu est caractéristique d’un syndrome paranéoplasique.
3 Causes
A Les neuronopathies ont pour causes principales :
- • le cancer du poumon à petites cellules dans le cadre d’un syndrome paranéoplasique (syndrome anti-Hu) : il convient de rechercher le cancer par les moyens les plus exhaustifs (scanner thoracique, biopsie, TEP-TDM); la recherche d’anticorps antineuronaux (dont ceux de type Hu) est systématique au cours de toutes les neuronopathies sensitives;
- • le syndrome de Gougerot-Sjögren : recherche de syndrome sec oculaire et buccal, biopsie des glandes salivaires accessoires, bilan biologique adapté;
- • une origine dysimmunitaire sans cause;
- • une origine toxique : sels de platine des chimiothérapies;
- • causes génétiques rares : CANVAS (Cerebellar Ataxia, Neuropathy, Vestibular Areflexia Syndrome), maladie de Friedreich, mutation POLG.
G 143Mononeuropathies multiples
A Elles correspondent à l’atteinte de plusieurs troncs nerveux. Le diagnostic est urgent afin de ne pas retarder le traitement d’une éventuelle vascularite nerveuse. Elles sont liées :
- • aux maladies de système : périartérite noueuse (vascularite), sarcoïdose, cryoglobuliné-mie (devant faire rechercher une infection par le virus de l’hépatite C);
- • au diabète (le plus souvent aux zones de compression des nerfs);
- • à la lèpre (neuropathie la plus fréquente dans le monde).
1 Clinique
- • A Dans un ou plusieurs territoires de troncs nerveux (fibulaire, médian, ulnaire) touchés par le processus ischémique.
- • Installation aiguë ou subaiguë.
- • Douleurs, paresthésies pénibles, brûlures.
- • Déficit sensitif puis moteur, évoluant vers l’amyotrophie.
- • Évolution extensive, confluente, prenant alors le masque d’une polyneuropathie car devenue symétrique.
- • L’atteinte des nerfs des membres est prédominante. Le nerf le plus fréquemment atteint est le nerf fibulaire commun.
- • L’atteinte des nerfs crâniens est possible (nerfs trijumeaux, nerfs oculomoteurs), en particulier au cours du diabète.
- • Le contexte général oriente vers une maladie de système : fièvre, amaigrissement, arthral-gies, myalgies et signes cutanés (livedo, purpura).
2 Examen neurophysiologique
L’examen neurophysiologique :
- • montre l’atteinte pluritronculaire axonale, asymétrique le plus souvent;
- • dépiste des anomalies infracliniques;
- • guide la biopsie sur le nerf sensitif le plus atteint pour la recherche d’une vascularite.
3 Biologie
À la recherche d’une cause :
- • NFS, VS, CRP, protéinurie des 24 heures;
- • sérologies : hépatites B et C, virus de l’immunodéficience humaine (VIH);
- • facteur rhumatoïde, C3, C4, CH50, cryoglobulinémie;
- • autoanticorps anti-noyaux, anticorps anti-ADN, anticorps anti-antigènes solubles (SSA, SSB, SM, centromères…), pANCA et cANCA.
4 Biopsie neuromusculaire
C’est l’examen clé pour la recherche d’une vascularite. Elle est réalisée sur le nerf sensitif le plus atteint, souvent le nerf fibulaire superficiel, branche cutanée du nerf fibulaire.
Elle peut montrer une vascularite nécrosante, avec nécrose fibrinoïde, un infiltrat inflammatoire de la paroi des artères de moyen calibre (fig. 8.17).
Coupe transversale de biopsie nerveuse : infiltrat inflammatoire de la paroi artérielle avec nécrose fibrinoïde. Coloration hématoxyline-éosine.
Image histologique en coupe transversale d’une biopsie nerveuse montrant un infiltrat inflammatoire dense autour de la paroi d’une artère intraneurale. L’organisation circulaire des cellules inflammatoires, à prédominance mononucléée, est clairement visible, entourant une lumière vasculaire altérée. La paroi artérielle présente une destruction structurelle marquée, avec une nécrose fibrinoïde bien identifiée par la zone centrale éosinophile homogène, traduisant une atteinte vasculaire sévère. À la périphérie, quelques adipocytes du tissu environnant sont reconnaissables, témoins du contexte périneural. L’ensemble de l’image illustre une vascularite active, caractérisée par une atteinte inflammatoire transmurale de la paroi artérielle et une nécrose fibrinoïde, typiques d’un processus pathologique auto-immun ou infectieux affectant les nerfs périphériques.
5 144Causes
- • Vascularites des vaisseaux moyens : périartérite noueuse.
- • Vascularites des petits vaisseaux :
-
- – vascularites à ANCA;
- – cryoglobulinémie essentielle.
- • Vascularites associées à une maladie de système :
-
- – cryoglobulinémie en rapport avec une infection par le virus de l’hépatite C;
- – lupus systémique;
- – infection par le VIH;
- – polyarthrite rhumatoïde (en régression).
- • Vascularites associées à une probable étiologie : vascularite satellite d’un cancer.
- • La périartérite noueuse peut se manifester par une atteinte isolée des nerfs périphériques.
- • La lèpre, responsable de mononeuropathies multiples à grande prédominance sensitive, est une grande cause de neuropathie dans le monde, mais rare en France.
- • Si le diabète est retenu comme cause à une mononeuropathie multiple, il n’y a pas de nécessité à réaliser une biopsie neuromusculaire.
6 145Traitement
Le diagnostic et donc le traitement des mononeuropathies multiples sont urgents, afin de limiter la perte en fibres nerveuses du nerf atteint par l’ischémie.
Le traitement repose notamment sur les corticoïdes et/ou les immunosuppresseurs.
7 Neuropathies motrices à blocs de conduction persistants
La neuropathie motrice à blocs de conduction persistants est une mononeuropathie multiple motrice pure, dysimmunitaire.
a Clinique
- • Elle touche habituellement l’homme adulte de moins de 50 ans.
- • Début asymétrique, prédominant aux membres supérieurs, touchant un ou plusieurs troncs nerveux, en particulier le nerf radial.
- • Déficit moteur, fasciculations, amyotrophie dans les territoires atteints (signes d’atteinte du neurone moteur périphérique).
b Diagnostic
L’ENMG montre des blocs de conduction, en dehors des zones de rétrécissement anatomique, persistants à 3 mois d’intervalle, sur les troncs nerveux touchés. Le degré de perte axonale témoigne de l’évolutivité de l’atteinte.
c Biologie
Présence d’anticorps anti-GM1 de type IgM dans la moitié des cas.
d Traitement
Il repose sur les immunoglobulines polyvalentes intraveineuses. Les corticoïdes peuvent aggraver les signes.
e Diagnostic différentiel
Essentiellement la sclérose latérale amyotrophique (voir ci-dessous).
II Distinguer cliniquement une neuropathie périphérique et une sclérose latérale amyotrophique
A Pour comprendre la sclérose latérale amyotrophique
La sclérose latérale amyotrophique (SLA), ou maladie de Charcot, ou maladie de Lou Gehrig aux États-Unis, est une affection motrice pure, liée à un processus dégénératif lésant le neurone moteur central (ou motoneurone central ou premier motoneurone) et le neurone moteur périphérique (ou motoneurone périphérique ou deuxième motoneurone) dans le territoire bulbaire et spinal.
146
Le pronostic est caractérisé par une évolution constamment fatale : la médiane de survie est de 36 mois en l’absence de traitement.
Cette affection se traduit par une paralysie des muscles striés (motricité volontaire) et respecte la sensibilité et la musculature lisse innervée par le système sympathique et parasympathique.
B Diagnostic
1 Diagnostic clinique de la SLA
a Principe du diagnostic
La SLA se caractérise cliniquement par un déficit moteur pur, progressif, combinant un syndrome du motoneurone périphérique (MNp) avec un syndrome du motoneurone central (MNc) dans le territoire bulbaire, cervical et lombosacré. L’âge moyen du début de la maladie se situe entre 60 et 65 ans.
b Éléments clés du diagnostic
Le diagnostic de SLA repose essentiellement sur les données cliniques et l’examen neurophysiologique. Les explorations complémentaires ont plus pour finalité d’exclure les pathologies qui peuvent mimer une SLA.
Pour le diagnostic d’une SLA, il faut une atteinte du neurone moteur central caractérisé par un syndrome pyramidal.
Compte tenu de l’atteinte simultanée des systèmes nerveux centrale et périphérique, les modifications des réflexes tendineux sont variables.
La présence de troubles cognitifs n’exclut pas le diagnostic : en effet, près de 20 % des patients atteints de SLA développent des troubles cognitifs de types frontotemporaux. Cela se traduit par des troubles du comportement et du langage, des changements dans la personnalité des patients, des difficultés mnésiques de type sous-corticales. De véritables associations SLA et démence frontotemporales sont possibles.
2 Diagnostic positif
A Le diagnostic de la SLA se fait à l’aide de l’ENMG. Il permet de confirmer l’atteinte de la corne antérieure, de préciser l’étendue des lésions et d’exclure une atteinte des troncs 147nerveux, en particulier une neuropathie motrice multifocale à blocs de conduction persistants (NMMBC), qui est le diagnostic différentiel principal.
Le diagnostic de SLA sera renforcé par la constatation :
- • d’une diminution des amplitudes des potentiels d’action moteurs et des vitesses de conduction normales ou peu ralenties;
- • de l’absence de blocs de conduction évocateurs d’une NMMBC lors de la stimulation des troncs moteurs;
- • de la normalité de l’exploration neurophysiologique sensitive;
- • de l’enregistrement de tracés de détection combinant un tracé de dénervation active au repos (potentiels de fibrillations, potentiels lents de dénervation et potentiels de fascicula-tions) et des tracés neurogènes faits de potentiels d’unité motrice (PUM) polyphasiques de grande amplitude, dont la fréquence s’élève avec l’effort;
- • de l’impossibilité de la systématisation de ces anomalies qui ne respectent aucun territoire radi-culaire, tronculaire et plexique et qui dépassent un territoire radiculaire, tronculaire et plexique.
Tout comme l’examen clinique, la normalité de l’exploration sensitive neurophysiologique est un élément clé du diagnostic.
C Diagnostics différentiels d’une SLA
1 Distinguer une SLA d’une neuropathie
À un stade précoce de la SLA, l’atteinte motrice peut prédominer sur le NMc ou de façon plus déroutante sur le NMp, conduisant alors à une confusion possible avec une neuropathie périphérique.
- • B Le diagnostic différentiel le plus complexe et le plus important est celui d’une NMMBC dont la présentation clinique peut mimer une SLA. À la différence de la SLA, l’âge de début est habituellement plus précoce avec une prédominance masculine et une propension à une atteinte initiale asymétrique des membres supérieurs. À la différence de la SLA, le territoire moteur déficitaire est tronculaire (un nerf médian et un nerf radial, par exemple) et non métamérique, le territoire bulbaire est épargné, l’évolution plus lente et il n’y a aucun signe d’atteinte du neurone moteur central. Comme dans la SLA, l’atteinte sensitive est absente. L’élément le plus discriminant est l’ENMG qui objective des blocs de conduction sur les troncs moteurs et une distribution tronculaire de l’atteinte. La présence d’anticorps anti-GM1 de type IgM renforcera le diagnostic. La prescription régulière de cures d’immunoglobulines intraveineuses permet une récupération motrice objective.
- • Les syndromes tronculaires étagés (syndrome du canal carpien et/ou du tunnel ulnaire) sont reconnus sur l’existence de troubles sensitifs, le caractère systématisé du déficit neurologique et les données de l’ENMG.
- • Les neuropathies motrices post-radiques sont le plus souvent constatées aux membres inférieurs. Elles surviennent dans un délai pouvant varier de 3 mois à 20 ans suivant l’irradiation. L’installation du déficit dans un territoire irradié et la présence de myoky-mies lors de l’examen ENMG confortent le diagnostic.
- • Les neuropathies secondaires à une intoxication au plomb doivent également être évoquées devant un déficit « pseudo-radial » des mains qui « font les cornes » (lors de l’extension des doigts, seuls l’index et l’auriculaire s’étendent à la différence de l’annulaire et du majeur qui restent fléchis). L’existence de douleurs abdominales et d’une anémie renforce cette hypothèse.
- • L’amyotrophie bulbo-spinale liée à l’X ou syndrome de Kennedy est une maladie génétique associée à des répétitions de triplets nucléotidiques CAG du gène des récepteurs aux andro-gènes. Elle touche l’homme adulte entre 30 et 40 ans et correspond à une perte progressive du MNp. Elle est responsable d’une atteinte motrice proximale touchant particulièrement la face et la langue. Cette atteinte bulbaire est source de dysarthrie et dysphagie et s’accompagne de 148fasciculations. Le déficit des membres, proximal, prédominant aux membres inférieurs est plus tardif. L’insensibilité aux androgènes est responsable d’atrophie testiculaire et de gynécomastie.
2 Pathologies musculaires et de la jonction neuromusculaire
- • A Les formes bulbaires de SLA et, de façon plus rare, les formes débutant par un déficit des extenseurs du rachis cervical peuvent conduire à suspecter une myasthénie. Cependant, l’atteinte oculomotrice caractéristique de la myasthénie est absente dans la SLA. À l’inverse, le diagnostic de myasthénie est porté par l’examen ENMG, objectivant une atteinte jonctionnelle neuromusculaire et la positivité du titre anticorps antirécepteurs de l’acétylcholine (++).
- • La myosite à inclusions est l’une des myopathies acquises de l’adulte les plus fréquentes. Il faut évoquer ce diagnostic devant tout tableau de SLA dont l’évolution est très lente ou nulle et réaliser alors une biopsie musculaire. Classiquement, le déficit moteur prédomine sur les quadriceps et les fléchisseurs du carpe.
- • A Les myopathies font partie des diagnostics différentiels :
-
- – parmi elles, prédominent les dystrophies des ceintures et les dystrophies myotoniques de type II, les maladies métaboliques comme la maladie de Pompe, le déficit en carnitine et les myopathies inflammatoires comme la polymyosite et les maladies mitochondriales.
- –
Item 97 – Polyradiculonévrite aiguë inflammatoire (syndrome de Guillain-Barré)
- I 149Définition
- II Épidémiologie et physiopathologie
- III Aspects cliniques
- IV Examens complémentaires
- V Traitement
- VI Surveillance et pronostic
Synonyme de syndrome de Guillain-Barré (SGB) : polyradiculonévrite inflammatoire aiguë.
I Définition
- • On désigne sous le terme de polyradiculonévrite aiguë une neuropathie périphérique inflammatoire, démyélinisante, sensitivo-motrice touchant les racines nerveuses et les nerfs les prolongeant.
- • On désigne sous le terme de neuropathie motrice axonale aiguë (acute motor axonal neuropathy ou AMAN) une neuropathie périphérique inflammatoire aiguë, médiée par des anticorps ciblant les nœuds de Ranvier des nerfs myélinisés, responsable d’une atteinte motrice pure aiguë.
II Épidémiologie et Physiopathologie
- • A On estime l’incidence à 1 pour 100 000.
- • Dans plus de 50 % des cas, le SGB est précédé d’un épisode infectieux respiratoire ou digestif : cytomégalovirus (CMV), virus d’Epstein-Barr (formes démyélinisantes) et diarrhées à Campylobacter jejuni (formes axonales de type AMAN).
-
• Les lésions touchent les racines des nerfs et les troncs nerveux en aval (fig. 8.18). Plusieurs racines et leurs prolongements nerveux sont lésés simultanément, possiblement de façon diffuse (les quatre membres et la face) sous la forme de lésions de démyélinisation.
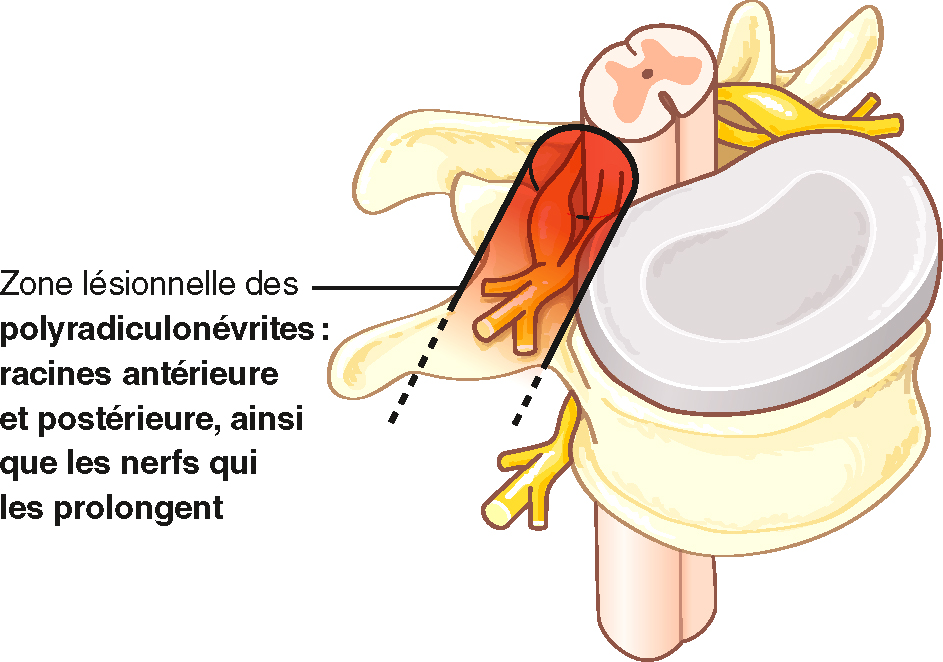
Fig. 8.18 Polyradiculonévrites aiguës sensitivo-motrices : localisation des lésions.
Illustration de Carole Fumat.Les expressions anatomiques représentent une vue latérale de la racine latérale qui se produit par rapport au foramen vertébral et aux disques intervertébraux adjacents à travers le foramen vertébral dans la moelle épinière. La zone rouge met l'accent sur l'emplacement des lésions observées dans une inflammation pulmonaire multiple avec des motifs très sensibles, affectant à la fois le moteur avant de tir et la racine postérieure sensible et les nerfs périphériques. Le voyage radiculaire des racines vertébrales met l'accent sur le parcours radiculaire vers la fusion dans les nerfs de la colonne vertébrale, indiquant la nature diffuse de l'implication inflammatoire. Les photographies soutiennent que la zone pathologique ne se limite pas au point d'origine, mais s'étend à plusieurs structures du système nerveux périphérique. Cela explique le mouvement bilatéral et les symptômes sensibles qui sont souvent observés dans ces attaques. Ce système favorise une compréhension topographique des dommages neuroinflammatoires dans le contexte de syndromes tels que Gilanbale.
III 150Aspects cliniques (vidéo 8.4)
- • On différencie deux types de SGB :
-
- – le SGB démyélinisant sensitivo-moteur avec risque d’intubation;
- – les neuropathies motrices axonales aiguës (AMAN), motrices pures, secondaires aux diarrhées à Campylobacter jejuni, habituellement sans risque d’intubation.
- • Les deux types évoluent en trois phases : extension, plateau, récupération.
- • Le diagnostic est porté sur la clinique et l’ENMG.
- • L’examen du LCS et l’immunologie sont d’un appoint non indispensable au diagnostic, mais utile pour éliminer les diagnostics différentiels (méningoradiculites).
A Phase d’extension des paralysies
- • La phase d’extension dure par définition moins de 4 semaines, mais elle est souvent de quelques jours (et peut être inférieure à un jour).
-
• Les manifestations sont variées (fig. 8.19) :
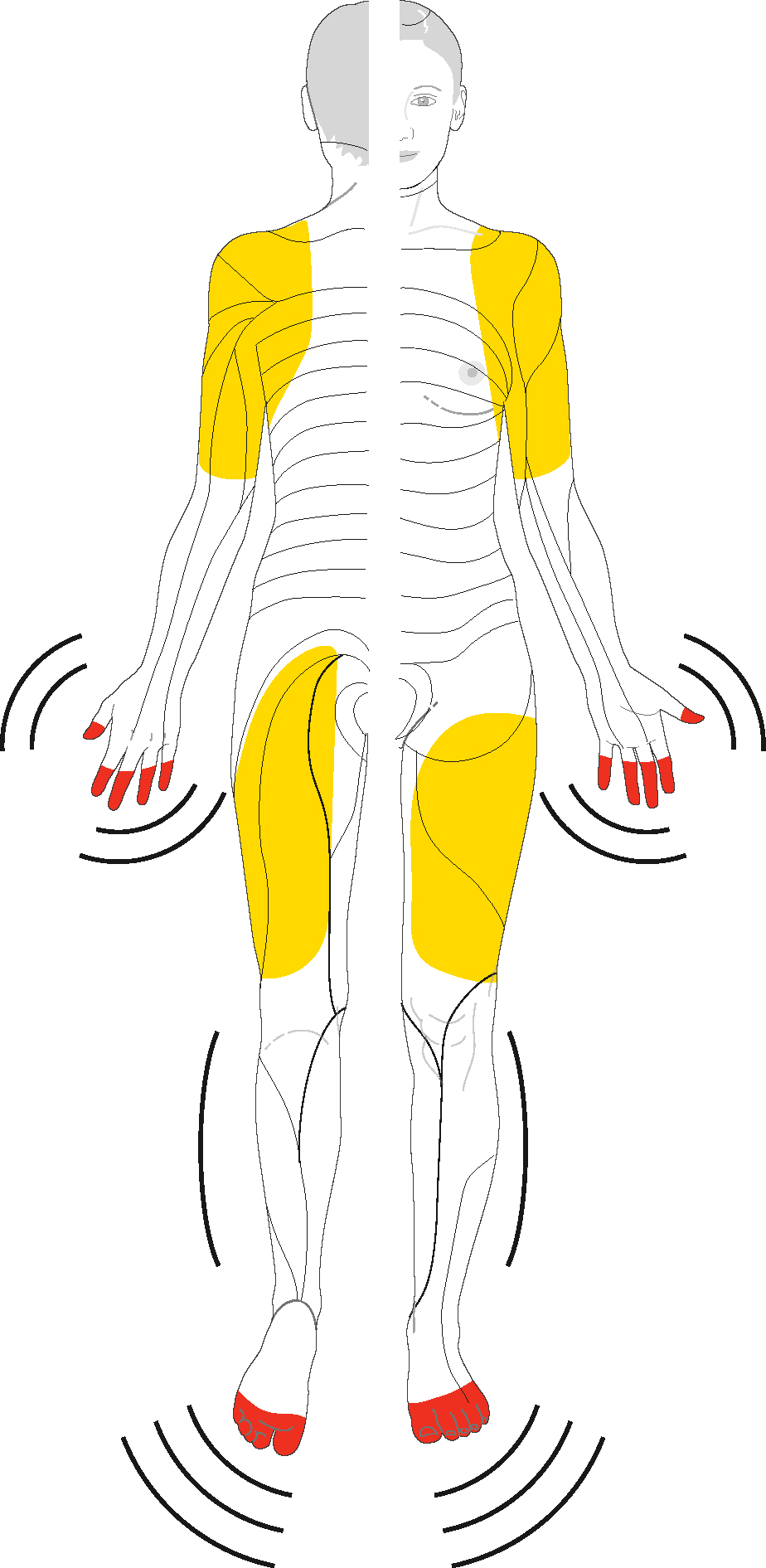
Fig. 8.19 Présentation clinique des polyradiculonévrites aiguës sensitivo-motrices (syndrome de Guillain-Barré).
En jaune : déficit moteur; en rouge : déficit sensitif; traits noirs : ataxie.
Illustration de Carole Fumat.Illustration anatomique représentant la répartition des symptômes dans les formes sensitivo-motrices aiguës des polyradiculonévrites, notamment dans le syndrome de Guillain-Barré. La figure montre un corps humain vu de face, avec une coloration en jaune localisée aux épaules, aux bras proximaux, aux cuisses et au tronc, traduisant une atteinte motrice des racines nerveuses et des nerfs périphériques. Les extrémités des mains et des pieds sont marquées en rouge, soulignant des troubles sensitifs distaux typiques comme les paresthésies. Des ondes noires en arc de cercle symbolisent les perceptions anormales comme les picotements ou engourdissements. Cette répartition illustre l’atteinte ascendante et symétrique classique du syndrome.
-
- – sensitives fréquentes et plutôt subjectives (paresthésies, picotements distaux des quatre membres), à évolution ascendante;
- – parésie débutant aux membres inférieurs, touchant rarement les nerfs crâniens (facial, oculomoteur, dysphagie) à la phase initiale; l’atteinte du cou et du tronc est plus tardive;
- – douleurs (myalgies, radiculalgies des membres inférieurs évocatrices).
- • L’atteinte motrice est une parésie relativement symétrique, étendue et sévère, qui prédomine en proximal puis touche les extrémités.
- • La gravité de l’atteinte respiratoire (15 à 30 % des patients seront sous ventilation assistée) impose une surveillance attentive en réanimation dès aggravation ou atteinte de la musculature bulbaire (troubles de déglutition ou de phonation).
- • Une atteinte faciale bilatérale et une aggravation rapide sont associées à un risque plus élevé de détresse respiratoire.
- • Une durée courte de la phase d’aggravation dans les formes démyélinisantes est de mauvais pronostic.
- • La phase d’extension est plus rapide au cours de l’AMAN qu’au cours du SGB démyélinisant.
B 151Phase de plateau
- • Classiquement :
-
- – un tiers des patients garde une capacité à marcher;
- – un tiers est confiné au lit;
- – un tiers nécessite une assistance respiratoire.
- • 152Le déficit moteur est d’intensité variable. L’atteinte des nerfs crâniens est fréquente : nerf facial (souvent diplégie) et troubles de déglutition (derniers nerfs crâniens), alors que l’atteinte des nerfs oculomoteurs est plus rare.
- • L’aréflexie tendineuse dans les territoires déficitaires est la règle.
- • Le déficit sensitif prédomine sur la proprioception et est responsable d’ataxie.
- • L’atteinte du système nerveux végétatif est fréquente (tachycardie, hypotension orthostatique, anomalie de la sudation, constipation) dans les formes sévères.
- • La durée du plateau est variable, plus longue dans les formes sévères (jusqu’à plusieurs mois) et dans certaines AMAN.
C Phase de récupération
- • La récupération se fait habituellement dans l’ordre inverse de l’apparition des déficits.
- • Au cours du SGB démyélinisant, elle peut durer plusieurs mois.
- • Au cours de l’AMAN, la récupération est soit rapide par levée des blocs de conduction sous traitement par immunoglobulines IV, soit très lente sur plusieurs mois en cas de persistance des blocs de conduction distaux et de dégénérescence axonale.
- • L’absence de récupération après 12 à 18 mois peut être considérée comme définitive.
- • À l’issue de la maladie, 5 % des patients sont décédés et 15 % gardent des séquelles définitives, notamment déficit moteur ou ataxie.
IV Examens complémentaires
A Aspects électrophysiologiques
1 Au cours du syndrome de Guillain-Barré démyélinisant
- • Les anomalies sont habituellement retardées par rapport à la clinique.
- • Initialement, allongement de la latence des ondes F et des latences distales en rapport avec l’atteinte radiculaire et distale.
- • À la phase d’état, anomalies démyélinisantes (évaluées sur les nerfs moteurs) caractérisées par une augmentation de la latence distale motrice (atteinte des fibres les plus rapides), ralentissement des vitesses de conduction, blocs de conduction, dispersion des potentiels.
- • Il n’y a pas de parallélisme entre le degré de la paralysie et les anomalies constatées en début d’évolution.
- • L’examen de détection met en évidence des anomalies neurogènes.
- • Seule l’inexcitabilité des nerfs est de mauvais pronostic.
2 Au cours de l’AMAN
- • Il n’existe pas d’anomalies sensitives.
- • Les amplitudes des potentiels moteurs sont diminuées aux quatre membres.
- • La détection montre des tracés neurogènes.
- • Il n’y a pas d’anomalie démyélinisante.
B 153Liquide cérébrospinal
- • A Son étude permet de mettre en évidence une dissociation albumino-cytologique caractérisée par :
-
- – une hyperprotéinorachie, pouvant être supérieure à 1 g/L, retardée de 3 à 10 jours par rapport au début de la clinique; elle peut atteindre plusieurs grammes par litre. Il n’y a pas de parallèle entre l’évolution clinique et l’importance de l’hyperprotéinorachie;
- – une absence de réaction cellulaire (< 10 mm3).
- • Une pléiocytose supérieure à 50 mm3 doit faire évoquer un autre diagnostic, telle qu’une méningoradiculite infectieuse.
C Autres examens biologiques
- • La biologie standard peut mettre en évidence :
-
- – une lymphopénie;
- – des anomalies du bilan hépatique (augmentation des transaminases).
- • La pratique des sérologies n’a qu’un objectif épidémiologique : CMV, virus d’Epstein-Barr; il faut penser à la séroconversion VIH (cependant rare).
- • En cas de diarrhée : sérologie Campylobacter jejuni.
- • Immunologie : les anticorps antigangliosides anti-GM1 et/ou anti-GD1a de type IgG sont associés aux AMAN.
V Traitement
A Traitements spécifiques
- • Le traitement doit être le plus précoce possible, au mieux dans les deux premières semaines.
- • Deux modalités sont possibles :
-
- – les immunoglobulines polyvalentes : à la dose de 0,4 g/kg par jour pendant 5 jours consécutifs en IV sur plusieurs heures;
- – les échanges plasmatiques : quatre échanges au total qui seront réalisés un jour sur deux.
- • Ces deux options thérapeutiques sont d’efficacité équivalente. Les immunoglobulines sont plus utilisées car de réalisation plus simple.
- • L’efficacité de ces traitements est prouvée sur la réduction de la durée de ventilation assistée, la rapidité de reprise de la marche et la durée d’hospitalisation.
- • Ces traitements n’ont pas modifié le pourcentage de patients avec séquelles.
- • L’association échanges plasmatiques et immunoglobulines polyvalentes est inutile.
- • Les corticoïdes sont inutiles.
B Traitements symptomatiques
- • Prévention indispensable des complications de décubitus : héparinothérapie par héparine de bas poids moléculaire, prévention des positions vicieuses favorisées par les déficits.
- • Ventilation mécanique si nécessaire.
- • Contrôle de la dysautonomie.
- • 154Rééducation précoce et poursuivie pour lutter contre la désadaptation à l’effort.
- • Mesures sociales pour l’aide à la reprise du travail.
VI Surveillance et pronostic
A Surveillance
1 Lors de la phase d’aggravation
- • Dépistage des troubles de déglutition.
- • Dépistage de difficultés respiratoires : efficacité de la toux, rythme respiratoire, mesure de la capacité vitale par spiromètre portable ou du débit expiratoire de pointe.
2 Lors de la phase d’état
- • Troubles végétatifs : modifications du rythme cardiaque, variation tensionnelle.
- • Complication de décubitus : thrombose veineuse, positions vicieuses.
B Pronostic
- • Sont de mauvais pronostic :
-
- – une phase d’aggravation très rapide;
- – une atteinte faciale bilatérale initiale;
- – un âge supérieur à 60 ans;
- – une inexcitabilité des nerfs à l’ENMG;
- – une ventilation prolongée.
- • Les récidives du SGB sont très rares.
 155Compléments numériques
155Compléments numériques
Des compléments numériques sont associés à ce chapitre. Ils proposent des vidéos indiquées dans le texte par un picto. Pour voir ces compléments, connectez-vous sur http://www.emconsulte/e-complement/478607 et suivez les instructions.
Vidéo 8.1. C Exemple d’exploration neurophysiologique : examen du nerf médian en électroneuro-myographie.
Vidéo 8.2. A Syndrome du canal carpien.
À l’interrogatoire, on retient la survenue au cours de la grossesse, l’atteinte bilatérale, le caractère douloureux et nocturne, la nécessité de bouger les doigts pour faire diminuer la douleur. On note également le caractère permanent des dyses-thésies et l’atteinte du 4e doigt. L’examen clinique confirme l’hypoesthésie et les dysesthésies dans les territoires symp-tomatiques. Les signes de Tinel et Phalen sont négatifs ou peu positifs. L’examen moteur n’est pas montré dans la vidéo.
Vidéo 8.3. B Neuronopathie sensitive.
On retient la symptomatologie initiale à type d’étau dans les membres inférieurs qui évoque une atteinte proprio-ceptive. L’extension des troubles aux membres supérieurs, également de type atteinte de la sensibilité profonde, est asymétrique, touchant d’abord la main droite avant la main gauche. Les troubles sont responsables d’une maladresse gênant l’écriture ou la manipulation des petits objets. Aux membres inférieurs, l’ataxie empêche de fermer les yeux sous la douche, de monter des escaliers ou de descendre une pente sans aide. La patiente précise bien qu’il ne s’agit pas d’un manque de force. Si la diffusion des membres inférieurs aux membres supérieurs dans un deuxième temps peut évoquer une polyneuropathie, l’atteinte ultérieure asymétrique et le caractère sensitif pur sont en faveur d’une neuronopathie sensitive. Enfin, à l’examen clinique, le signe de Romberg est positif, les réflexes tendineux sont tous abolis. Même si la pallesthésie n’est abolie qu’aux membres inférieurs, le diagnostic (confirmé par l’électroneuromyogramme) est celui de neuronopathie sensitive.
Vidéo 8.4. A Polyradiculonévrite aiguë.
On retient chez cette patiente la survenue à la suite d’une infection respiratoire, de douleurs radiculaires très sévères, continues, touchant d’abord les membres inférieurs puis les membres supérieurs. Elles sont caractéristiques du syndrome de Guillain-Barré et sont prises dans l’exemple pour des douleurs d’origine mécanique. On note également leur apparition aux membres supérieurs. Par la suite surviennent des troubles sensitifs des pieds et des jambes ainsi que des mains. La patiente se plaint aussi de lourdeur des membres inférieurs qui correspondent vraisemblablement à un déficit moteur. Enfin, la paralysie faciale reflète le caractère diffus de l’atteinte neurologique. La patiente montre la déviation de la bouche vers la droite en raison de la paralysie faciale gauche. À noter que la patiente signale que son œil gauche avait tendance à se fermer. Il s’agit vraisemblablement d’une atteinte du muscle frontal responsable de la chute de la queue du sourcil, à ne pas confondre avec l’orbiculaire de l’œil dont le déficit est responsable de la mauvaise occlusion de l’œil dans la paralysie faciale. Il faut également entendre le caractère orienté de l’interrogatoire qui permet de reconstituer les différentes étapes d’une polyradiculonévrite : événement déclenchant, déficit sensitif des quatre membres, puis déficit moteur avec paralysie faciale.
10 = Pas de contraction. 1 = Contraction faible sans déplacement. 2 = Déplacement faible si pesanteur éliminée. 3 = Déplacement possible contre pesanteur. 4 = Déplacement possible contre résistance. 5 = Force normale; la réponse idiomusculaire est conservée.